


Summary
L-DOPA를 사용하여 도파민 대체 약물은 파킨슨병에서 가장 일반적으로 사용되는 증상을 치료하지만, 불수의 이상 운동, 되나 운동 이상증을 포함한 부작용이 동반된다
Abstract
든 이미징 질량 분석법 (IMS)은 생물 학적 조직 샘플 2 (Fig.1)의 분자 종의 공간적 분석을 용이하게하는 강력한 방법이다. 12 μm의 얇은 조직 섹션은 일반적으로 든 TOF / TOF 질량 분석기를 사용하여 질량 분석기로 감지할 수 온전 펩티드와 단백질의 탈착 및 이온화를 촉진 든 행렬로 덮여 있습니다. 일반적으로 피크 수백은 단일 쥐의 뇌 조직 섹션에서 평가 할 수 있습니다. 일반적으로 사용되는 영상 기술 달리, 이러한 방식은 관심 분자의 사전 지식을 필요로하지 않습니다 높은 분자 특이성과 감도 2를 유지하면서 여러 분자 종 혼자 가잖아하고 종합적인 분석을 허용합니다. 여기에서 우리는 동물 모델 파킨슨병 (PD)의 쥐의 두뇌에 neuropeptides의 특정 지역과 관련된 유통 프로필을 elucidating위한 든 IMS 기반의 접근 방식을 설명합니다.
PD 나이 3,4 65 명 이상 1 %의 유행과 함께 일반적인 neurodegenerative 질병입니다. 가장 일반적인 증상 치료는 L-DOPA 5를 사용 도파민 교체에 따라 달라집니다. 그러나 이것은 L-DOPA 유발 dyskinesias (뚜껑) 1,3,6 되나 불수의 이상 운동을 포함한 심각한 부작용이 동반된다. 뚜껑에서 가장 두드러진 분자 변화 중 하나는 opioid 전구체 prodynorphin의 mRNA 7 upregulation이다. dynorphin의 펩티드는 본질적으로 운동 제어 7,8에 관련된 뇌 영역에서 neurotransmission 보죠. 그러나, neuropeptide 전구체의 처리에서 발생한 정확한 opioid 펩티드의 연대를 특징되지 않았습니다. 따라서, 우리는 실험 파킨슨병 및 L-DOPA 유도 운동 이상증의 동물 모델에서 든 IMS를 활용.
든 이미징 질량 분광법은 neuropeptide characteriza에 관하여 특히 유리한 것으로 판명기, 일반적으로 사용되는 항체 기반의 접근 방식부터 알려진 펩타이드 시퀀스와 이전에 관찰 포스트 translational 수정을 대상으로하고 있습니다. 반대로 든 IMS는 새로운 펩타이드 가공 제품을 해명하기 때문에의 연결을 전송 neuropeptide 변조의 새로운 분자 메커니즘을 밝힐 수 있습니다. neuropeptides의 절대 액수가 든 IMS에 의해 결정될 수 없지만 펩타이드 이온의 상대적인 풍요로움은 건강과 질병의 레벨 변경에 대한 통찰력을 제공, 대량 스펙트럼에서 delineated 수 있습니다. 여기에 제시된 예제에서는 dynorphin B, 알파 neoendorphin과 물질 P의 정상 농도는 크게 dorsolateral 증가된다는 것을 발견했다지만, 심한 운동 이상증과 동물의 dorsomedial, striatum는 (그림 얼굴, 몸통과 orolingual 근육을 포함하지 5). 또한, 든 IMS는 기능성 inacti의 이전에 알려지지 않은 메커니즘을 대표하는, 운동 이상증의 심각도 및 DES-티로신 알파 neoendorphin 수준 간의 상관 관계를 밝혀N-터미널 티로신의 제거 등 striatum에서 dynorphins의 vation는 dynorphin의 opioid - 수용체 결합 능력 9를 줄여줍니다. 이 든 IMS와 결과가 생물 의학 연구의 모든 분야에 응용 프로그램에 대한 기술의 잠재력을 강조를 사용하여 뚜껑의 neuropeptide 특성에 관한 최초의 연구이다.
Protocol
프로토콜은 일반적으로 여러 쥐의 뇌 섹션, 20-30 섹션에서 든 IMS 데이터의 통계 분석의 목적에 맞게 변경하고, 조직 준비로 구성된 다섯 가지 단계로 구성되어 있으며, 매트릭스 응용 든-TOF MS 분석, 데이터 평가 및 neuropeptide 신분증. 절차에 설명된 아래 상세 설명되어 있습니다 :
1. 조직 준비
이 절차는 각 조직 샘플의 수집뿐만 아니라 IMS 분석 sectioning 조직을 포함합니다. 단백질과 펩타이드 분석의 특정 목표는 proteolytic 저하를 방지하는 것입니다. 따라서 그것은 조직 해부하는 동안 신속하고 부지런을 작동하는 것이 필수적이다.
- 잘린으로 쥐 (일반적으로 250-300그램)를 희생, -80 ° C 냉동고에 전송하기 전에 가루 드라이 아이스의 <의 30 대 및 동결의 최대 사후 시간 내에 쥐의 두뇌를 제거합니다. 액체 질소를 사용하여 빠른 동결부정적인 매트릭스 결정에 영향을줍니다 뇌 조직에서 microtears의 위험을 증가함으로써 (그림 2D) MS의 품질을 줄일 수 있습니다. 전체 두뇌는 석사 신호 품질의 손실없이 sectioning 전에 몇 년 동안 저장할 수 있습니다.
- 12 μm의의 조각과 해동 마운트 조직 섹션 전도성 든 유리 슬라이드에 (인듐 주석 산화물 코팅 슬라이드, Bruker Daltonics) 또는 든 대상 (그림 2A-C)에 그라 이오 스탯의 마이크로톰에 냉동 조직을 잘라.
- -80 15 진공 이하 분 및 매장 슬라이드를위한 드라이 섹션 ° C 이상 사용까지. 조직 섹션은 -80에 저장된 경우에도 sectioning 후 가능한 최단 시간 내에 분석해야 ° C. 우리는 MS 신호 품질이 눈에 띄게 스토리지 년 후 줄어들는 것을 발견했습니다. 단백질과 펩티드의 산화를 줄이기 위해서는, 저장 용기에 공기를 불활성 가스 (예 : 아르곤 또는 질소)로 교체할 수 있습니다.
2. 매트릭스 응용
매트릭스 응용단계는 스펙트럼의 품질에 큰 영향을 가지고 있으며, 조직의 종류뿐만 아니라 관심 analyte에 따라 여러 매개 변수의 최적화가 필요합니다. 이러한 요소는 모체의 종류, 매트릭스 농도, 산도, 조직 세척 및 유기 수정자뿐만 아니라 예금 볼륨, 선서 10 수평 해상도와 숫자 (그림 2D)를 포함하여 기기 설정과 같은 화학 매개 변수를 포함합니다. 대규모 실험의 경우는 하루 만에 모든 부분에 매트릭스를 적용하여 같은 연산자에 의해 예를 들어, 편차를 줄이기 위해 매우 중요합니다. 이러한 승화하거나 스프레이, 작은 매트릭스 방울의 배열의 자동화 증착에 의한 크기 100-150 picoliter에 대해 다양한 조직의 작은 단백질과 neuropeptides의 분석에 성공적으로 사용되었습니다대로 행렬 솔루션을 적용할 수있는 많은 전략이 있지만 , 뇌 섹션 9, 10,11, 12, 13 포함합니다.
- 한 H에 대한 dessicator에 서리를 없애다는 섹션우리.
- 실험이 운영자가 아닌 다른 사람의 눈을 멀게되었는지 확인합니다. 다시 레이블 모든 샘플.
- 10 초 동안 70 %의 에탄올 (EtOH, 상온에서, RT)에 섹션 1X 씻고 두번의 95% 10 초위한 EtOH (RT). 대형 실험 들어, 모든 유리 세탁을 수행 편차를 최소화하기 위해 cuvette을 사용하여 함께 슬라이드.
- 10 분 대한 dessicator의 섹션을 건조합니다.
- 현미경 조직 부분을 평가하고 든 MS 품질 (그림 2D)에 악영향을 줄 것이다 조직 왜곡, microtears와 작은 균열이 있는지 확인합니다.
- 50 % 메탄올 10 % 150mM 암모늄 아세테이트 (AmAc)와 물 0.3 % trifluoroacetic 초산 (TFA)에서 50 밀리그램 / ML DHB 구성된 신선한 매트릭스 솔루션을 준비합니다.
- 매트릭스 응용 프로그램은 화학 잉크젯 프린터를 사용하여 사각형 패턴 (칩, Shimadzu)에 이산 비말 증착에 의해 수행됩니다. 첫 번째 단계는 neuropeptide 분석 includi위한 행렬 응용 프로그램의 실험 매개 변수를 최적화하는 것입니다패스 당 방울의 NG 번호, 패스의 수. 이 실험은 각 배열과 같은 영장의 callosum, 피질 및 striatum와 유사한 두뇌 영역을 다루는되어 있는지 확인하는 반면, 동일한 조직 섹션에서 다른 응용 프로그램 매개 변수를 여러 매트릭스 배열을 적용하여 수행됩니다. 같은 실험 때마다 매개 변수가 서로 다른 뇌 구조, 특정 analytes을 대상으로 서로 다른 매트릭스, 그리고 다른 매트릭스 용제는 특정 analytes의 추출에 필요한 경우. 포함하여 변경 수행되어야한다
- 조직 섹션에있는 유리 슬라이드 홀더를 스캔하고 홀더에 맞춥니다. 조직 섹션에 매트릭스 응용 프로그램에 대한 귀하의 배열을 정의하고 거리를 탐지하는 공간적 해상도 즉 장소를 지정합니다. 화학 잉크젯 프린터에 최적화된 프로토콜을 사용하여 행렬을 적용합니다. 본 실험에서는 우리가 다음과 같은 인쇄 매개 변수를 가진 펩타이드 이미징을위한 최적화된 프로토콜 사용 : 10 방울 (100 PL / 드롭), 10 응용 프로그램은 패스와 D를 탐지하는 장소300 μm의의 istance.
- 스캔 최종 매트릭스는 섹션을 발견하고 사전에 든 데이터 수집 (3.4 단계)에 등록을 사진을 저장합니다.
- 진공 하에서 dessicator에 더 이상 사용할 때까지 섹션을 저장합니다.
3. 든 MS 데이터 수집 및 처리
neuropeptides의 MS 분석은 모든 단일 매트릭스 명소 14에서 소프트웨어를 이용한 데이터 수집을 사용 항공기구 (Ultraflex II, Bruker Daltonics, 독일) 반사 모드에서 작동을 든 시간에 수행됩니다. 따라서 정확한 공간적 학습이 필수적입니다. 그것은 든 최적화, 인수 특히 대상 등록 실험 선호 실험 그룹 멀게해야 동일한 연산자에 의해 수행되는 필수적이다. 다른 사람이 화학 잉크젯 프린터를 운영하는 동안 여러 유리 슬라이드와 함께 대규모 실험에 든 실험은 한 운영자에 의해 수행할 수 있습니다.
- 질량 분석계에 유리 슬라이드를로드합니다.
- 낮은 분자량 표준 교정 믹스 (Bruker Daltonics)를 사용 든 수집 방법의 교정을 확인합니다.
- 인수 매개 변수를 최적화합니다.
- MS 신호를 최적화하고 인접 행렬 예금에서 매트릭스를 흡열 방지하기 위해 조직에 레이저와 최적의 초점의 크기가 결정되어야합니다.
- 레이저 에너지는 최대 해상 도나 포화 탐지기를 줄이고 기준을 앞질렀어 않고 가능한 많은 매트릭스 예금 등에서 최대 MS의 품질을 향상시키기 위해 설정됩니다.
- 전용 소음이 감지되기 전까지 행렬 스폿 당 촬영의 최대 개수를 평가, 자주 1000년에서 2000년까지 촬영. 축적된해야하고 현장 내에 레이저 위치를 전에 얻은 촬영의 수를 변경해야 촬영의 수를 예상됩니다. 균등하게 각 행렬 자리를 샘플링하기 위해, 우리는 도망을 사용하여 24 단계의 총 번호, 25 촬영 단계에서 600 총소리를 축적각 매트릭스 증착에서 운동의 DOM 패턴.
- FlexImaging를 사용 든 단계의 모터 좌표 (v.2.0) 10에 대한 모든 발견 섹션 스캔을 등록하고 AutoXexuteBatchRunner.exe 소프트웨어를 사용하여 배치 모드에서 데이터 수집을 수행합니다.
- 기준 뺄셈 (볼록 선체 V3), 스무딩 및 ASCII 파일 (*. DAT, *. txt 또는 *. csv 형식)으로 수출이어서 외부 보정 (옵션)에 의해 각각의 단일 스펙트럼을 처리합니다. 15
4. 데이터 평가
최종 자료 평가는 통계 분석이어서 피크 내용에 초점을하여 데이터 후처리 및 데이터 감소로 구성됩니다.
- 초기 단계로 든 IMS 섹션은 overnormalization 효과에 대해 평가했다. 이것은 쉽게 그러한 FlexImaging (Bruker Daltonics) 또는 BioMap (노바티스)와 같은 데이터 시각화 도구를 채용하여 얻을 수 있습니다. 초기 단계로 총 이온 이미지 에바 있습니다총 이온 전류 (TIC) 정상화하기 전에 luated 여러 저명한 펩티드 봉우리 단일 이온 분포 이미지의 수동 검사로 따라갔다. 특성 피크 강도 배포판을 찾아보고 그들이 품질 또는 정상화 효과를 (그림 3) 스포팅, 조직 기능 (손해 배상)에 관련하는 경우.
- histological 기능에 따라 관심 지역 (striatum을 예) 윤곽을 그리다 및 ASCII 파일 형식의 해당 스펙트럼을 내보냅니다. 가급적, 총 이온 전류 (TIC)에 스펙트럼의 정상화는이 단계에서 수행할 수 있습니다.
- 같은 기원과 같은 데이터 처리 소프트웨어 (v.8.1, Originlab), MATLAB (MathWorks, Natick, MA, 미국) 또는 R 16 ASCII 파일을 가져옵니다. 피크 검출은 예를 기원이나 Matlab에서 "mspeaks"의 "피크 분석"을 위해, 소프트웨어에 포함된 피크 찾기 도구를 사용하여 수행할 수 있습니다. 하나의 탭으로 구분된 텍스트 파일로 모든 스펙트럼에서 peaklists를 내보냅니다.
- 검색된 펩티드를 위해 빈 테두리를 결정하기 위해서는봉우리, binning 분석은 적절한 소프트웨어 도구를 (예 : pbin 17)를 사용하여 수행하거나 자체 여기 MATLAB 또는 R.위한 스크립트를 작성 모든 피크를 포함하는 단일 텍스트 파일의 데이터를 선택한 소프트웨어에로드되어 피크 국경 결정에 대한 매개 변수가 지정되어있다 피크는 실험에 대한 관련 되려면 스펙트럼에 명시되어야 할 빈도 등. 예를 들어, 실험 동물 2 그룹, 각 그룹에 5 동물, 100 스펙트럼이자 각 동물 및 지역으로부터 수집되는 포함되어 있습니다. 그것이 한 그룹 (3 / 5)과 그 동물의 스펙트럼 (3x50 = 150 스펙트럼) 중 적어도 절반의 동물 중 최소한 과반수에있는 경우 피크가 잠재적으로 흥미 롭다고 가정하고,이 전체 비율에게 제공합니다 총 1000 (2x5x100) 스펙트럼의 150 긍정적인 스펙트럼 밖으로 15 %를 사용합니다. pbin 도구를 사용하여이 단계에서 얻은 데이터에서 결정된 모든 빈 폭을 포함하는 단일 binrange 파일을 듭니다. 그 빈 테두리를 확인할 수 있도록적절한이며, 그것은 원래의 스펙트럼 트레이스와 함께 오리진의 방식을 시각화하기 쉽습니다.
- 피크 - 지역 통합은 통계 분석을 위해 중요하다 편차를 줄일 수 있습니다. 우리는 4 단계에서 결정된 정상 테두리 사이의 곡선 아래 면적을 계산하는 연구를 위해 자체 작성한 스크립트를 사용합니다. 통합 피크 영역은 18 번 수행되는 SAM 도구를 사용하여 비 파라메 트릭 unpaired 테스트를 통해 MS 엑셀 (v.2007) 및 통계 분석으로 가져옵니다.
5. 펩타이드 식별
관찰된 펩타이드의 정체성 시퀀스 확인은 생물 학적 타당성을 체결하기 위해서는 필수적입니다. 높은 펩타이드의 농도는 분석 12,13 이런 종류의 필요한 있지만 가장 정확한 방법은 직접 탠덤 질량 분석법 (MS / MS)에 의해 펩타이드 조각을 사용하여 조직 오프 진정한 하향식 결정이 포함됩니다. 낮은 풍부 펩티드 또는 clos있는 여러 펩티드를 위해그놈 / Z 값 (± 0.5 %), 온 - 조직 분석은 장애되고 peptidomic 전략을 사용하여 해제 조직 분석 추출, 분리 및 내생 neuropeptides의 MS 기반의 식별을 포함하는 활용됩니다. 여기에 제시된 실험 내용은 중앙 초점이 펩티드는 스펙트럼의 다른 neuropeptides에 비해 풍부한 매우 낮을 때문에 특정 도전입니다 opioid 펩타이드 감지,있었습니다. 일반적인 펩타이드 추출 및 분리 기술을 보유하는 것은 상대적으로 친수성과 상태가 말이 또한, 이러한 펩티드 오히려 극지 있습니다 .. 따라서 우리는 펩타이드 식별 9,19을 기반으로 표준 LC-MS/MS와 함께 조직 추출 및 opioid 펩타이드 prefractionation에 대한 이전보고 프로토콜을 적용했습니다.
- 관심 대상 구조 (; 꼬리가있는 putamen, CPU 핵 accumbens, NAC)의 코로나 부분을 수집합니다. 그라 이오 스탯의 마이크로톰에서 냉동 쥐의 두뇌를 탑재하고 (오호 주변 뇌 물질을 제거텍스, 때론 심장, 코퍼스의 callosum). 해부하는 NAC와 CPU와 해동 NAC와 다양한 유리 슬라이드에있는 섹션의 CPU 부품을 탑재의; 섹션 (N = 50 30 μm의)를 수집합니다.
- 2 분 동안 품어, 100 μL 5퍼센트 ACN/0.1 %의 TFA를 추가하여 조직 오프 펩티드를 추출하고 eppendorf 낮은 단백질 바인딩 튜브에 수집합니다. 이 단계를 두 번 반복합니다.
- 증가 이온 강도를 19에서 stepwise (N = 4) 용리를 사용하여 강력한 양이온 교환 크로마 토그래피에 의해 펩타이드 prefractionation을 수행합니다. speedvac 농축기를 사용하여 진공 하에서 시료를 건조합니다.
- 전기 분무 탠덤 질량 분광법 (LC-MS/MS)에 인터페이스 nanoflow C18 반대 위상 액체 크로마 토그래피의 방법 (1100, 애질런트 테크놀로지, 산타 클라라, CA)에 의해 펩티드 분수를 분석. 석사 실험 하이브리드 선형 iontrap / 푸리에 변환 이온 사이클로 트론 공명 (FTICR) 악기 (LTQ FT 7T, 써모 과학, Waltham, MA), 펩타이드 fulscan 스펙트럼 (M / Z 150에서 수행되었다-2000)는 충돌 유발 분해 (CID) 9 의한 iontrap에서 5 가장 강렬한 펩티드 봉우리의 후속 조각이어서 높은 질량 분해능 (100000)에 FTICR 분석기와 함께 인수했다.
- 펩타이드 서열 식별은 데이터베이스 검색에 의해 수행되며, 드 노보 시퀀싱 분석하여 보완 할 수 있습니다. 데이터베이스 검색의 경우 상용 검색 엔진은 (마스코트, XTandem 또는 단백질 광부) 20을 고용하고 있습니다. 검색은 일반적으로 알려진 또는 예측 neuropeptides 및 neuropeptide 전구체 단백질 21의 시퀀스의 시퀀스를 포함하는 데이터베이스에 대해 수행됩니다.
6. 대표 결과
약 300 monoisotopic 분자 종 (평균 스펙트럼은 그림에 해당하는 1000 개 이상의 봉우리 검색 결과 여기에서 설명한 절차에 따라 준비로 striatal 조직 섹션 든 이미징 질량 분광법그림 인치 1). 저명한 분자 이온 봉우리를위한 데이터 시각화는 플렉스 이미징 소프트웨어를 사용 달성 및 해부 학적 특징 (Fig.3)에 맞춰 잘 지내지 특성 피크 강도 배포판을 보여줬습니다. 든 IMS의 추가 기능은 상대적으로 좋은 재현성입니다. 본 실험에서 검색된 모든 분자 종의 정상 농도에 대한 분산의 전반적인 계수 30 % 였지만 많은 봉우리가 매우 낮은 변동 및 치료 그룹 내에서 높은 재현성 (그림 4) 표시됩니다. lesioned하고 그대로 둘 striata의 dorsolateral 및 dorsomedial 일부를 포함하여 관심있는 네 가지 영역의 상대적인 피크 강도 데이터를 통계 분석에 노출되었다. 동시에 여러 비교를위한 조정하기 위해 통계 분석은 SAM 도구 18 사용 nonparametric 테스트에 의해 수행되었다. 가장 눈에 띄는 변경 사항은 도파민-denervated, parkinsonian striatum의 dorsolateral 부분에서 발견되었다. 여기에시 dynorphin B와 알파 neoendorphin (Fig.5), 다른 치료 그룹 사이에 gnificant 변경은 두 dynorphin의 펩티드를 위해 관찰되었다. 세부에서 50~60% 의한 dynorphin의 피크 농도 모두 상대적인 증가는 낮은 dyskinetic 동물과 병변 컨트롤 (에 비해 높은 dyskinetic 동물에서 관찰되었다 P <0.05, F (2, 15) = 12.8 DynB, F = 5.7 aNeo; 그림. 5).

. 꼬리가있는 putamen (CPU)와 핵 accumbens (NAC), 그림 1은 평균 MS는 striatum의 두 밀접하게 관련된 영역에서 얻은 는거죠. 두 지역은 고유 한 영역으로 표현 몇 가지 분자 여종의 서로 다른 MS 프로파일을 표시하거나, 다른 피크 강도 수준에서 (삽입, M / Z 2028). 각 피크의 공간적 분포 패턴은 전문 이미징 소프트웨어 (하단 패널)을 사용하여 시각하실 수 있습니다.
"2.jpg />
그림 2 (A) 두뇌는 퍼가기 미디어 (OTC, 화살표)를 사용 그라 이오 스탯 척에 마운트됩니다., OTC는 펩티드의 OTC 원인 이온 억제 이후 sectioned 될 수있는 뇌 영역을 오염하지 않는다는주의 가져옵니다. (B, C) 얇은 섹션 (≈ 12 μm의 두께) 육안으로 탐지하기 어려울 수 있습니다 든 호환되는 유리 슬라이드에 해동 마운트와 C (D) Microtears에서 본대로 동결 피해를 방지하기 위해 몇 초 동안 건조 하지만 든 매트릭스 결정에 악영향을하며 든 MS 신호를 완전히 쓸어 버릴 수 있습니다. cresyl 보라색 물들일 같은 섹션 microtears 및 균열 (오른쪽 하단 현미경 사진)을 보여준다.

그림 3. 데이터 평가의 첫 번째 단계 (AI) 분석 질량 범위에 걸쳐 여러 봉우리를 시각화하는 것입니다. 여기, 9 생쥐에서 striatal 섹션이 든 MS와 함께 몇 군데 있었다. 평균 총 시각화 전현재에 눈에 높거나 낮은 이온 농도 (화살표)의 영역을 보여줄 것입니다. 이 영역 이상 또는 이하 - 정상화 효과와 결과를 손상 왜곡 데이터 분석에 의해 영향을받을 수 있습니다. 피크 배포판 나쁨 해부 학적 정의는 나를 통해, 예를 들어 섹션 3, 9, 일반적으로 낮은 피크 신호 대 잡음과 봉우리의 F를 섹션을 표시

그림 4. 치료 그룹 사이의 MS 재현성은 평균 MS 추적하고 각 M / Z 값 (삽입, M / Z 722 및 1749)에 대한 표준 오차를 계산하여 평가 할 수 있습니다. 좋은 재현성의 유효 기간은 통계 분석을 보장합니다.
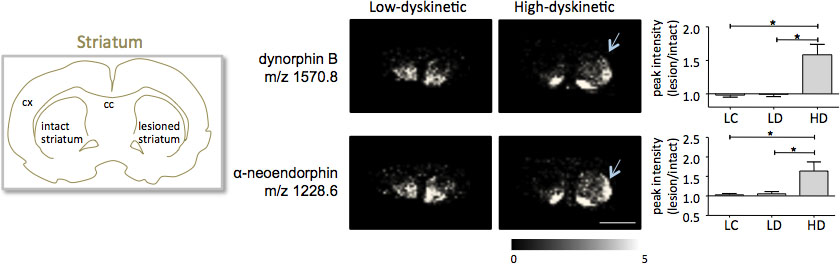
그림 5. Dynorphin B와 알파 neoendorphin 피크 농도는 크게 6-OHDA에 증가하고 있습니다- lesioned, parkinsonian 높은 dyskinetic 동물의 striatum (HD, 화살표)가 낮은 dyskinetic (LD)와 병변 제어 그룹 (LC)에 비해. 펩타이드 피크 농도는 그대로 측면의 평균 접이식 변화 ± SEM (그대로 / 병변 쪽)으로 표현. * P <0.05, CX 피질, CC 코퍼스의 callosum. 스케일 바 5mm.
Discussion
neuropeptides의 연구에 든 이미징 질량 분광법을 고용의 몇 가지 장점이 있습니다. MS 데이터의 편견 분석은 특정 뇌 핵을 표시하거나 같은 striatum의 단지 dorsolateral 부분은 특정 pathophysiological 조건과 연결된 곳이 여기 제시된 결과에 있습니다. 공간 정보를 유지함으로써 그것은 전체 뇌 부분의 분석 또는 펩타이드 추출물에서 전통 peptidomics 연구를 사용 비해 높은 감도와 낮은 다양성과 통계적 분석을 수행하기 위해 관심 영역을 재정의하는 데 후 가능합니다. 또한, 그것은 든 IMS는 즉시 이전에 알려지지 않은 사후 translational 수정을 감지할 수 실현하는 것이 중요하지만, 구조적 분석은 수정되어 정확한 아미노산 위치를 결정하기 위해 따라야합니다.
든 IMS 데이터를 시각화의 일반적인 함정은 BL에서 선형 광학 규모로 최대 피크 강도를 매핑 포함대신 100 % 모든 부분의 최대 피크 강도가 어디 흔한 절대 규모에 대한 모든 부분을 매핑의 실험 시리즈의 각각의 섹션에서 (그림 3)에 대한 컬러 (100 %)까지 (0 %) 응답 (그림 5) . 후자의 방법은 그룹 데이터 및 치료 그룹 사이의 차이의 시각화의 비교를 허용합니다.
든 IMS 분석의 주요 장애물은 특정 질량 봉우리에 펩티드의 과제입니다. 온 조직 탠덤 - 질량 분광법 때로는 가능하지만 종종 13,14 꽤 어려움이 증명한다. 우리는 반대 위상 LC-MS/MS 뒤에는 강력한 양이온 교환 크로마 토그래피, 대한 예비 분별 (분리)를 포함하여 더 전통적인 접근 방식이 성공적으로 시퀀스 많은 neuropeptides 특히 opioid 펩티드하는 데 사용할 수있는 것을 발견했습니다. 그것은 같은 마스코트와 같은 일반적인 검색 엔진을 사용하여 모든 데이터베이스 항목이 일치하지 않는 양질의 MS / MS 스펙트럼을 얻기 위해 여전히 흔치 않습니다. 이러한 경우에는 손으로 드 노보-시퀀싱은 onl입니다Y 옵션도 있습니다. 피크 정체성의 궁극적인 증명은 적절한 녹아웃 마우스에서 조직 섹션 든 IMS에 의해 얻을 수있다, 그러나 이것은 항상 사용할거나 불가능합니다. 대안은 서구 immunoblotting이나 immunohistochemistry에 의해 예를 들어, 정반대로 다른 방법으로 결과를 확인하는 것입니다. 이것은 종종 항체 및 신규 항체를 확인하는 작업을 상당히 높이는 포함될 수 있습니다.
이 프로토콜에 제시된 일반적인 전략은 몇 가지 섹션 및 실험 조건을 포함한 대규모 neuropeptide 든 IMS 실험에 최적화되어 있습니다. 메커니즘 밑에있는 고통과 중독의 약물에 대한 내생적인 반응을 포함하여 연구의 다양한 분야에서 취업으로 프로토콜은 특히 opioid 펩티드에 최적화되었으며, 향후 연구에 큰 영향을 미칠 것이다.
Disclosures
저자가 공개하는 게 없다.
Acknowledgments
우리는 소중한 의견을위한 그림 3과 교수 요나 Bergquist에 대한 데이터를 기여한 한나 워너 감사드립니다. 스웨덴 연구 협의회 (부여 522-2006-6416 (MA), 521-2007-5407 (MA), 아케 Wiberg의 재단 (MA, JH), 과학 로얄 스웨덴어 아카데미 (MA, JH), 그리고 스웨덴의 화학 사회 (JH)가 기꺼이 재정적인 지원을 인정한다.
References
- Obeso, J. A., Olanow, C. W., Nutt, J. G. Levodopa motor complications in Parkinson's disease. Trends Neurosci. 23, S2-S7 (2000).
- Caprioli, R. M., Farmer, T. B., Gile, J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. MALDI-TOF MS. Anal. Chem. 69, 4751-4760 (1997).
- Obeso, J. A. The evolution and origin of motor complications in Parkinson's disease. Neurology. 55, S13-S20 (2000).
- O, W. H. Noncommunicable Diseases and Mental Health Cluster, Noncommunicable Disease Prevention and Health Promotion Department, Ageing and Life Course. Active Ageing: A Policy framework. , (2002).
- Schapira, A. H. Movement disorders: advances in cause and treatment. Lancet Neurology. , 6-7 (2010).
- Obeso, J. A., Rodriguez-Oroz, M. C., Rodriguez, M., DeLong, M. R., Olanow, C. W. Pathophysiology of levodopa-induced dyskinesias in Parkinson's disease: problems with the current model. Ann. Neurol. 47, S22-S32 (2000).
- Cenci, M. A., Lee, C. S., Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 10, 2694-2706 (1998).
- Andersson, M., Hilbertson, A., Cenci, M. A. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis. 6, 461-474 (1999).
- Hanrieder, J. Alterations of striatal neuropeptides revealed by imaging mass spectrometry. Molecular & Cellular Proteomics. , (2011).
- Cornett, D. S., Reyzer, M. L., Chaurand, P., Caprioli, R. M. MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods. 4, 828-833 (2007).
- Ljungdahl, Imaging Mass Spectrometry Reveals Elevated Nigral Levels of Dynorphin Neuropeptides in L-DOPA-Induced Dyskinesia in Rat Model of Parkinson's Disease. PLoS ONE. 6, e25653 (2011).
- Groseclose, M. R., Andersson, M., Hardesty, W. M., Caprioli, R. M. Identification of proteins directly from tissue: in situ tryptic digestions coupled with imaging mass spectrometry. J. Mass. Spectrom. 42, 254-262 (2007).
- Andersson, M., Groseclose, M. R., Deutch, A. Y., Caprioli, R. M. Imaging mass spectrometry of proteins and peptides: 3D volume reconstruction. Nat. Methods. 5, 101-108 (2008).
- Deininger, S. -O. Imaging Mass Spectrometry. Setou, M. , Springer. Japan. 199-208 (2010).
- Norris, J. L. Processing MALDI Mass Spectra to Improve Mass Spectral Direct Tissue Analysis. Int. J. Mass. Spectrom. 260, 212-221 (2007).
- Ihaka, R., Gentleman, R. R. A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics. 5, 299-314 (1996).
- Mass Spectrometry Binning Software GAB. , Vanderbilt Center for Quantitative Sciences. Nashville, TN. Available from: http://www.vicc.org/biostatistics/software.php (2012).
- Tusher, V. G., Tibshirani, R., Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116-5121 (2001).
- Bergstrom, L., Christensson, I., Folkesson, R., Stenstrom, B., Terenius, L. An ion exchange chromatography and radioimmunoassay procedure for measuring opioid peptides and substance P. Life. Sci. 33, 1613-1619 (1983).
- Falth, M. Neuropeptidomics strategies for specific and sensitive identification of endogenous peptides. Mol. Cell. Proteomics. 6, 1188-1197 (2007).
- Falth, M. SwePep, a database designed for endogenous peptides and mass spectrometry. Mol. Cell. Proteomics. 5, 998-1005 (2006).
