

Summary
Analyses ARN de séquençage et de bioinformatique ont été utilisés pour identifier les facteurs de transcription de manière significative et exprimés de manière différentielle dans des sous-populations Lin-CD34 + et CD34 Lin-des EMLcells de souris. Ces facteurs de transcription peuvent jouer un rôle important dans la détermination de l'interrupteur entre les cellules Lin-CD34 auto-renouvellement Lin-CD34 + et partiellement différenciés.
Abstract
Les cellules souches hématopoïétiques (CSH) sont utilisés en clinique pour le traitement de la greffe à reconstruire le système hématopoïétique d'un patient dans de nombreuses maladies telles que la leucémie et le lymphome. Élucider les mécanismes contrôlant CSH auto-renouvellement et de différenciation est important pour l'application de CSH pour la recherche et les utilisations cliniques. Cependant, il est impossible d'obtenir des CSH en grande quantité en raison de leur incapacité à proliférer in vitro. Pour surmonter cet obstacle, nous avons utilisé une lignée de cellules dérivées de la moelle osseuse de souris, la lignée cellulaire EML (érythroïde, myéloïde et lymphoïde), en tant que système modèle pour cette étude.
ARN-séquençage (ARN-Seq) a été de plus en plus utilisés pour remplacer les puces à ADN pour les études d'expression génique. Nous rapportons ici une méthode détaillée de l'utilisation de la technologie de l'ARN-Seq pour enquêter sur les facteurs clés potentiels dans la régulation de la cellule EML auto-renouvellement et de différenciation. Le protocole fourni dans le présent document est divisé en trois parties. La première part explique comment la culture de cellules EML et séparé Lin-CD34 + et les cellules Lin-CD34. La deuxième partie du protocole propose des procédures détaillées pour la préparation d'ARN total et la construction de la bibliothèque ultérieure pour le séquençage à haut débit. La dernière partie décrit la méthode d'analyse de données RNA-Seq et explique comment utiliser les données pour identifier les facteurs de transcription exprimés de manière différentielle entre Lin-CD34 + et les cellules Lin-CD34. Les facteurs de transcription plus nettement exprimés de manière différentielle ont été identifiés comme les principaux régulateurs potentiels contrôle cellule EML auto-renouvellement et de différenciation. Dans la section de discussion de cet article, nous mettons en évidence les étapes clés de la performance réussie de cette expérience.
En résumé, ce document propose une méthode d'utilisation de la technologie de l'ARN-Seq pour identifier des régulateurs potentiels de l'auto-renouvellement et de différenciation dans les cellules EML. Les principaux facteurs identifiés sont soumis à l'analyse fonctionnelle en aval in vitro et in vivo.
Introduction
Les cellules souches hématopoïétiques sont des cellules sanguines rares qui se trouvent principalement dans la niche de la moelle osseuse adulte. Ils sont responsables de la production de cellules nécessaires pour reconstituer le sang et le système immunitaire 1. Comme une sorte de cellules souches, cellules souches hématopoïétiques sont capables à la fois de l'auto-renouvellement et de différenciation. Mécanismes qui contrôlent la décision de sort de CSH élucider, soit vers l'auto-renouvellement ou de la différenciation, offrira de précieux conseils sur la manipulation de cellules souches hématopoïétiques pour les recherches de maladies du sang et de l'utilisation clinique 2. Un problème rencontré par les chercheurs est que les CSH peuvent être maintenues et développées in vitro dans une mesure très limitée; la grande majorité de leur progéniture sont partiellement différenciées en culture 2.
Afin d'identifier les principaux régulateurs qui contrôlent les processus d'auto-renouvellement et de différenciation à l'échelle de l'ensemble du génome, nous avons utilisé une primitive hématopoïétique ligne de cellules souches de souris EML en tant que système modèle. Thest la lignée cellulaire a été dérivée à partir de 3,4 murin de la moelle osseuse. Lorsque nourris avec différents facteurs de croissance, les cellules peuvent se différencier en EML érythroïdes, myéloïdes, lymphoïdes et des cellules in vitro 5. Surtout, cette lignée cellulaire peut être propagé en grande quantité dans le milieu de culture contenant un facteur de cellules souches (SCF) et en conservant leur multipotentialité. EML cellules peuvent être séparés en sous-groupes d'auto-renouvellement Lin-SCA + CD34 + et les cellules différenciées partiellement Lin-SCA-CD34 sur la base de marqueurs de surface CD34 et SCA 6. Similaire à court terme CSH, SCA + CD34 + cellules sont capables d'auto-renouvellement. Lorsqu'ils sont traités avec SCF, Lin-SCA + des cellules CD34 + peut régénérer rapidement une population mixte de Lin-SCA + et les cellules CD34 + Lin-SCA-CD34 et continuent à proliférer 6. Les deux populations sont similaires dans la morphologie et des niveaux similaires de c-kit ARNm et la protéine 6. Des cellules Lin-SCA-CD34 sont capables de se propager dans un milieu contenant IL-3 à la place de SCF 3. Unveiling régulateurs clés dans la décision du destin cellulaire EML à offrir une meilleure compréhension des mécanismes cellulaires et moléculaires à transition précoce de développement au cours de l'hématopoïèse.
Afin d'étudier les différences moléculaires sous-jacents entre le Lin-SCA auto-renouvellement + CD34 + et les cellules Lin-SCA-CD34 partiellement différenciées, nous avons utilisé l'ARN-Seq pour identifier les gènes exprimés de manière différentielle. En particulier, nous nous concentrons sur les facteurs de transcription, des facteurs de transcription jouent un rôle crucial dans la détermination du destin cellulaire. ARN-Seq est une approche développée récemment qui utilise les capacités de séquençage de nouvelle génération (NGS) technologies de profil et de quantifier les ARN transcrits à partir de 7,8 génome. En bref, l'ARN total est le poly-A et fragmenté choisi comme modèle initial template.The ARN est ensuite converti en ADNc en utilisant la transcriptase inverse. Afin de cartographier des transcrits d'ARN de pleine longueur, en utilisant intact, l'ARN non dégradé pour la construction de la banque d'ADNc est importante. Pour le purpose de séquençage, des séquences d'adaptation spécifiques sont ajoutées aux deux extrémités de l'ADNc. Ensuite, dans la plupart des cas, les molécules d'ADNc sont amplifiés par PCR et séquences d'une manière à haut débit.
Après séquençage, résultant lectures peut être aligné sur un génome de référence et une base de données de transcriptome. Le numéro de la carte qui lit le gène de référence est prise en compte et cette information peut être utilisée pour estimer le niveau d'expression du gène. Le lit peut également être assemblé de novo sans un génome de référence, ce qui permet l'étude de transcriptome dans les organismes non-modèles 9. la technologie de l'ARN-seq a également été utilisé pour détecter les isoformes d'épissage 10 à 12, de nouveaux transcrits 13 et 14 des fusions de gènes. En plus de la détection de gènes codant pour des protéines, de l'ARN-Seq peut également être utilisé pour détecter et analyser nouveau niveau d'ARN non codants, tels que la transcription de l'ARN de long 15,16, 17 micro-ARN, etc. siRNA 18 non codante. En raison de til précision de cette méthode, il a été utilisé pour la détection de variations nucléotidiques simples 19,20.
Avant l'avènement de la technologie de l'ARN-Seq, puces à ADN est la principale méthode utilisée pour analyser le profil d'expression génique. Sondes pré-synthétisées sont conçus et ensuite fixés à une surface solide pour former une lame de microréseau 21. L'ARNm est extrait et converti en ADNc. Au cours du processus de transcription inverse, les nucleotides marqués par fluorescence sont incorporés dans l'ADNc et l'ADNc peuvent être hybridés sur les lames de puces à ADN. L'intensité du signal recueilli à partir d'un endroit précis dépend de la quantité d'ADNc de liaison à la sonde spécifique à cet endroit 21. Par rapport à la technologie de l'ARN-Seq, puces à ADN a plusieurs limites. Tout d'abord, puces à ADN repose sur la connaissance préalable de l'annotation des gènes, tandis que la technologie de l'ARN-Seq est capable de détecter de nouveaux relevés de notes au niveau de fond élevé relative, ce qui limite son utilisation lorsque geniveau d'expression ne est faible. En outre, la technologie de l'ARN-Seq a beaucoup plus dynamique portée de détection (8000 fois) 7, tandis que, en raison de fond et la saturation des signaux, l'exactitude des puces à ADN est limitée pour les deux gènes fortement exprimés et humble 7,22. Enfin, des sondes de puces à ADN diffèrent dans leur efficacité d'hybridation, ce qui rend les résultats moins fiables lors de la comparaison des niveaux d'expression relatifs des différents produits de transcription au sein d'un échantillon 23. Bien que l'ARN-Seq a de nombreux avantages par rapport aux puces à ADN, l'analyse des données est complexe. Ceci est une des raisons pour lesquelles de nombreux chercheurs utilisent encore microarray la place de l'ARN-Seq. Divers outils de bioinformatique sont nécessaires pour le traitement et l'analyse des données 24 ARN-Seq.
Parmi plusieurs séquençage de nouvelle génération (NGS), les plates-formes 454, Illumina, Torrent SOLIDE et Ion sont les plus largement utilisés. 454 a été la première plate-forme commerciale NGS. A la différence des autres plates-formes de séquençagelongueur comme Illumina et solide, la plate-forme 454 génère plus lus (moyenne 700 BASE lit) 25. Plus de lectures est mieux pour la caractérisation initiale de transcriptiome en raison de leur plus assembler efficacité 25. Le principal inconvénient de la plate-forme 454 est son coût élevé par mégabase de séquence. Le Illumina et solides plates-formes de générer lit avec une augmentation du nombre et de courtes longueurs. Le coût par mégabase de séquence est beaucoup plus faible que la plate-forme 454. En raison du grand nombre de court interprète pour l'Illumina et solides plates-formes, l'analyse des données est beaucoup plus de calculs. Le prix de l'instrument et des réactifs pour le séquençage de la plate-forme Ion Torrent est moins cher et le temps de séquençage est plus courte de 25. Cependant, le taux d'erreur et le coût par mégabase de séquence sont plus élevés par rapport à la Illumina et les plates-formes solides. Différentes plates-formes ont leurs propres avantages et inconvénients et nécessitent des méthodes d'analyse des données. Le platform devraient être choisis en fonction du but de séquençage et la disponibilité des fonds.
Dans cet article, nous prenons plate-forme Illumina ARN-Seq comme un exemple. Nous avons utilisé cellule EML en tant que système modèle pour étudier les régulateurs clés dans EML cellule auto-renouvellement et de différenciation, et a fourni une des méthodes détaillées de la construction de la bibliothèque de l'ARN-Seq et l'analyse de données pour le calcul du niveau d'expression et roman détection de transcription. Nous avons montré dans notre précédente publication de cette étude de l'ARN-Seq dans EML système modèle 2, lorsqu'il est couplé avec un test fonctionnel (par exemple shRNA de knockdown) fournir une approche puissante dans la compréhension du mécanisme moléculaire des premiers stades de la différenciation hématopoïétique, et peut servir de modèle pour l'analyse de cellules auto-renouvellement et de différenciation en général.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML culture cellulaire et la séparation des cellules Lin-CD34 + et CD34-Lin en utilisant la cellule magnétique Système et tri cellulaire activé par fluorescence méthode de tri
- Préparation de rein de bébé hamster (BHK), du milieu de culture de cellules pour la collecte de facteur de cellules souches:
- Culture des cellules BHK dans un milieu DMEM contenant 10% de FBS en flacon de 25 cm2 (voir le tableau 1) à 37 ° C, 5% de CO2 dans un incubateur de culture cellulaire.
- Lorsque les cellules se développent à 80 - 90% de confluence, laver les cellules une fois avec 10 ml de PBS. Ajouter 5 ml de solution de trypsine-EDTA à 0,25% de la monocouche et on incube les cellules pendant 1-5 min à température ambiante (TA) jusqu'à ce que les cellules sont décollées.
- Pipet la solution et doucement à briser des amas de cellules. Ajouter 5 ml de DMEM complet dans le ballon pour arrêter l'activité de la trypsine. Recueillir les cellules par centrifugation à 200 xg pendant 5 min à température ambiante.
- Enlever le milieu et remettre en suspension le culot cellulaire dans 10 ml de milieu BHK fraîches de culture cellulaire. Transférer 2 ml de la suspension cellulaire de l'étape 1.1.4 dans un nouveau flacon de 75 cm 2 et ajouter 48 ml de milieu de culture de cellules BHK fraîches dans le ballon.
- Culture des cellules BHK pour deux jours et recueillir le milieu de culture. Passage du milieu à travers un filtre de 0,45 um. Conserver dans le milieu à -20 ° C jusqu'à utilisation ultérieure.
- EML culture cellulaire:
- Culture des cellules en suspension (EML) en milieu basique EML BHK contenant le milieu de culture des cellules (tableau 1) à 37 ° C, 5% de CO2 dans un incubateur de culture cellulaire.
- Maintenir les cellules EML à faible densité cellulaire (0,5 à 5 x 10 5 cellules / ml) avec la densité de crête inférieure à 5 x 10 6 cellules / ml. Diviser les cellules tous les 2 à 3 jours, à raison de 1: 5. Passage cellules EML doucement et jetez la culture après repiquage depuis 10 générations.
- L'épuisement des cellules positives de la lignée:
- Récolter les cellules EML par centrifugation à 200 xg for 5 min et laver les cellules une fois avec du PBS. Recueillir les cellules par centrifugation à 200 g pendant 5 min.
- Reprendre les cellules avec du PBS et compter les cellules avec un hémocytomètre. Déterminer la concentration d'anticorps dans l'étape de séparation ultérieure des cellules en fonction du nombre de cellules (se il vous plaît consulter les instructions proposées par le fournisseur du système d'isolement des cellules).
- Isoler la lignée négative (Lin-) cellules en utilisant un cocktail d'anticorps de la lignée (cocktail d'anticorps monoclonaux conjugué à la biotine CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G / C), 7-4 et Ter-119 ) et un système magnétique de tri cellulaire activé selon les instructions du fabricant.
- Séparation des cellules Lin-CD34 + et CD34-Lin:
- Isoler les cellules Lin- de l'étape 1.3.3 à 200 g pendant 5 min. Remettre en suspension le culot de cellules avec du PBS et compter les cellules avec un hémocytomètre.
- Laver les cellules deux fois avec un tampon FACS et sédimenter les cellules à 200 xgpendant 5 min.
- Étiqueter cinq 1,5 ml microtubes avec le numéro 1, 2, 3, 4, 5 respectivement. Reprendre les cellules avec 100 pi de tampon FACS pour 10 6 cellules (10 6 cellules par tube).
- Ajouter 1 pg d'anticorps anti-souris CD34 FITC dans le tube 1 et tube 2 et mélanger doucement les tubes.
- Incuber tous les tubes à 4 ° C pendant 1 heure dans l'obscurité.
- Ajouter 0,25 pg d'anticorps anti-Sca1 PE-conjugué et 20 pl d'APC conjugué anticorps lignée de cocktail de tube 1, 0,25 pg d'anticorps anti-Sca1 conjugué à PE de tube 3, et 20 ul d'anticorps lignée cocktail de APC conjugué à tube 4.
- Mélanger tous les tubes doucement et incuber les cellules à 4 ° C pendant encore 30 minutes dans le noir.
- Ajouter 300 ul de tampon FACS pour les cellules et centrifuger les cellules à 200 g pendant 5 min.
- Laver les cellules avec 500 pl de tampon FACS à trois reprises.
- Remettre en suspension le culot cellulaire dans 500 pi de FACS buffre.
- Utilisez les cellules dans des tubes 2, 3, 4, et 5 pour la mise en place de compensation. Isoler les cellules Lin-SCA + CD34 + et Lin-SCA-CD34 dans le tube 1 en utilisant FACS Aria.
2. Préparation d'ARN et construction de la bibliothèque de séquençage à haut débit
- L'isolement, la qualité de l'analyse et la quantification de l'ARN:
- Extrait de l'ARN total de Lin-CD34 + et les cellules Lin-CD34 en utilisant respectivement TRIzol suivant le protocole fabrique ».
- Retirer l'ADN en utilisant la désoxyribonucléase I contaminée (DNase I) selon le protocole du fabricant. Eventuellement, stocker l'ARN à -80 ° C à cette étape pour une utilisation ultérieure.
- Évaluer la qualité des ARN total en utilisant Bioanalyzer selon les instructions offerts par le fournisseur. Utilisez échantillon d'ARN avec l'ARN Nombre intégrité (RIN) de bière à 9.
- Construction d'une bibliothèque et séquençage à haut débit:
NOTE: Ce protocole décrit ARN-Seq en utilisant la plate-forme Illumina. Pourd'autres plates-formes de séquençage, différentes méthodes de préparation de bibliothèques sont nécessaires.- Utilisez 0,1-4 ug d'ARN total de haute qualité par exemple pour la préparation bibliothèque. Normalement 2 ug d'ARN total peut être extrait à partir de 10 5 cellules EML.
- Utiliser un système de préparation d'échantillons ARN-séquençage pour la purification d'ARN et de la fragmentation, premier et deuxième synthèse brin d'ADNc, réparation de fin, les extrémités 3 'adénylation, adaptateur ligature et l'amplification par PCR, en suivant les procédures standard détaillées par rapport aux instructions du fournisseur.
- Sélectionner positivement PolyA ARNm en utilisant oligo-dT billes magnétiques et fragmenter l'ARNm.
- Effectuer transcription inverse en utilisant des amorces aléatoires pour obtenir l'ADNc et ensuite synthétiser le deuxième brin d'ADNc pour générer de l'ADNc double brin.
- Retirez les surplombs »et remplir la 5 '3 surplombs par l'ADN polymérase. L'adénylate extrémités 3 'pour empêcher la ligature des fragments d'ADNc à partir d'une autre.
- Ajouter adaptateurs d'indexation multiplex aux deux extrémités de l'ADNc-bc. Effectuer la PCR pour l'enrichissement de fragments d'ADN.
- Mesurer la A260 / A280 pour obtenir des informations sur la concentration de bibliothèque à l'aide d'un spectrophotomètre.
- Évaluer la qualité de la bibliothèque et de mesurer la gamme de taille des fragments d'ADN en utilisant un Bioanalyzer.
3. Analyse des données
Pour référence des logiciels utilisés dans cette partie, se il vous plaît voir (tableau 2).
- le traitement des fichiers de données pour l'analyse en aval:
- Convertir .bcl (fichier d'appel de base) à déposer .fastq fichier en utilisant un logiciel de CASAVA (Illumina, la version 1.8.2).
- Lancez le 'Terminal' dans le système Linux. Allez dans le dossier de données qui contient le fichier de données à partir d'une machine de séquençage Illumina HiSeq2000. Supposons que le dossier de résultat est «NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX / ', le typedans la commande de la figure S1A, et entrer dans le dossier de données.
- Installez CASAVA 1.8.2 dans le système Linux. Supposons que le outputfolder est «non aligné», utilisez la commande à la figure S1B pour préparer le fichier de configuration pour la conversion. Utilisez l'option --fastq-cluster-comptage 0 à assurer un seul fichier .fastq est créé pour chaque échantillon. Le fichier généré est .fastq au format .gz. Décompressez-le pour l'analyse en aval (Figure S1B).
- Une fois le dossier 'Unaligned' a été généré, allez dans le dossier "Unaligned '(Figure S1C).
- Utilisez la commande de la figure S1D pour commencer le processus de conversion. Le paramètre '-j' fournit le numéro d'unité centrale de traitement qui sera utilisé.
- Après que le système a terminé le processus de conversion, allez dans le dossier de résultat sous dossier «non aligné» (Figure S1E).
- Utilisez la commande de la figure S1F </ Strong> pour décompresser le fichier .fastq.gz dans .fastq fichier sous chaque dossier de l'échantillon.
- Convertir .bcl (fichier d'appel de base) à déposer .fastq fichier en utilisant un logiciel de CASAVA (Illumina, la version 1.8.2).
- Détecter de nouveaux relevés de notes et d'évaluer le niveau d'expression en utilisant Tuxedo Suite 26:
- Plan de la fin de l'ARN-Seq couplé lit au génome de référence de la souris (UCSC version mm9, obtenu à partir de http://cufflinks.cbcb.umd.edu/igenomes.html ) en utilisant un logiciel de Tophat (version 1.3.3) 27, qui utilise la Bowtie lire mappeur (version 0.12.7) 28. Tophat est fourni avec l'option "-no-nouveaux-juncs" pour améliorer la précision de l'estimation du niveau de l'expression.
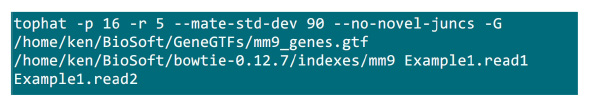
- Mettez les fichiers .fastq dans un dossier où le processus de cartographie sera mise en œuvre. Supposons qu'il y ait 2 fichiers (renommer .fastq à Example1.read1, Example1.read2) pour un échantillon de séquençage paired fin, utilisez la commande à la figure S2 pour faire la cartographie (ajuster les paramètres en fonction de la configuration du système).Le paramètre "-p" fournit le nombre de CPU qui sera utilisé. Les paramètres "-r" et "-mate-STD-dev" peuvent être obtenus à partir de la bibliothèque QC ou déduites à partir d'un sous-ensemble de lit alignés (Figure S2).
- Assemblez la cartographie lit en transcrits d'ARN en utilisant le logiciel de manchette (version 1.3.0) 29. Boutons de manchette analyse avec le fichier d'annotation de gènes connus (même fichier .gtf utilisé par Tophat) et le fichier .bam produite par Tophat.
- Après Tophat terminé la course, dans le même dossier, utilisez la commande à la figure S3A afin de fonctionner boutons de manchette pour construire transcriptome et transcription estimer le niveau d'expression. Le 'mm9_repeatMasker.gtf' et les fichiers de séquence du génome dans le dossier 'GenomeSeqMM9' peuvent être obtenus à partir du navigateur UCSC Genome.
- Les fichiers genes.expr et transcripts.expr obtenus contiennent la valeur de l'expression des gènes et des transcriptions (isoformes). Copier et collerle contenu du fichier dans un fichier Excel et de manipuler avec tableur (Figure S3B).
- Utilisez la commande de la figure S3C à comparer la «transcripts.gtf 'fichier à la référence« mm9_genes.gtf' fichier résultant afin d'identifier de nouveaux relevés de notes.
- Le fichier résultant contient .tmap le résultat de la comparaison. Copiez et collez le contenu du fichier dans un fichier Excel et manipuler avec tableur. Transcriptions avec le code de la classe «u» peuvent être considérés comme «roman» par rapport à la référence .gtf fichier fourni (Figure S3D).
REMARQUE: Pour l'analyse en aval commodité, définissez les valeurs de FPKM à 0,1 si les valeurs sont en dessous de 0,1.
REMARQUE: L'étape 3.2.3 - 3.2.6 est facultative pour ceux qui souhaitent améliorer la précision de l'estimation expression de nouveaux relevés de notes. Cela va prendre beaucoup plus de temps, parce que la cartographie et la construction du transcriptome doivent être rnon plus d'une fois.
- Exécutez Tophat en utilisant les paramètres par défaut et puis exécutez à boutons de manchette fichier .gtf généré à l'aide de la commande à la figure S3E.
- Comparez le fichier de .gtf résultant dans le fichier .gtf du génome de référence en utilisant la commande à la figure S3F.
- Analyser le fichier .tmap entraîné comme décrit à l'étape 3.2.2.4. Copiez et collez le contenu du fichier dans un fichier Excel et manipuler avec tableur. Transcriptions avec le code de la classe «u» peuvent être considérés comme «roman» par rapport à la référence .gtf fichier fourni.
- Après l'étape 3.2.5, il existe un fichier de .combined.gtf dans le dossier qui peut être utilisé en tant que fichier de .gtf de référence. Une deuxième série de boutons de manchette Tophat et peut être réalisée comme décrit dans l'étape 3.2.1 et 3.2.2 pour obtenir une estimation plus précise de FPKM de nouveaux relevés de notes.
- Plan de la fin de l'ARN-Seq couplé lit au génome de référence de la souris (UCSC version mm9, obtenu à partir de http://cufflinks.cbcb.umd.edu/igenomes.html ) en utilisant un logiciel de Tophat (version 1.3.3) 27, qui utilise la Bowtie lire mappeur (version 0.12.7) 28. Tophat est fourni avec l'option "-no-nouveaux-juncs" pour améliorer la précision de l'estimation du niveau de l'expression.
- Détecter differentially exprime des gènes en utilisant DESeq paquet de 30.
- L'entrée de DESeq est une brute tableau des chiffres de lecture. Pour obtenir un tel tableau, utilisez le script htseq comptage distribué avec le paquet HTSeq Python qui peut être téléchargé à partir du site HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Assurez-vous que samtools, python, et htseq comptage programsare installés dans le système. Obtenir des chiffres bruts de comptage de lecture de sortie de tophat en utilisant la commande à la figure S4A.
- Préparer, des fichiers "ExperimentDesign.txt 'aide d'Excel' Raw_Count_Table.txt. Copiez et enregistrez le contenu au format .txt pour le paquet DESeq R (Figure S4B).
- Installez programme de R dans le système. Dans le terminal, de type «R» et appuyez sur ENTER.A message sur écran appearas montré à la figure S4C.
- Lire «Raw_Count_Table.txt »,« ExperimentDesign.txt 'dans R en utilisant la commande à la figure S4D.
- Chargez paquet DESeq aide de la commande à la figure S4E.
- Conditions factoriser dans R (Figure S4F).
- Utilisez la commande de la figure S4G de réaliser des essais binominal négatif sur la table de comptage normalisée.
- Utilisez la commande de la figure S4H de gènes exprimés sortie différentiels importants dans un fichier .csv.
- L'entrée de DESeq est une brute tableau des chiffres de lecture. Pour obtenir un tel tableau, utilisez le script htseq comptage distribué avec le paquet HTSeq Python qui peut être téléchargé à partir du site HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Recherche des facteurs de transcription »(TFS) valeurs de FPKM à travers les échantillons en utilisant Excel. Intersection DE Table de gène et la table TF. Les gènes appartiennent à la fois à la table sont exprimés de manière différentielle des facteurs de transcription.
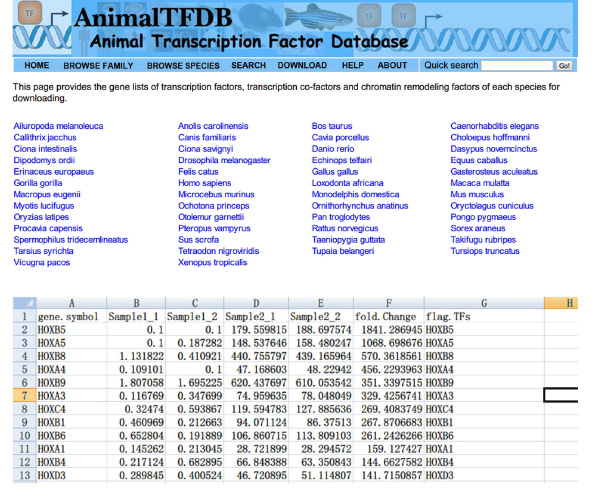
- Allez sur le site http://www.bioguo.org/AnimalTFDB/download.php et télécharger les facteurs de transcription. Cherchez ensuite les facteurs de transcription de Dans le Excel (< strong> Figure S5).
- Génération fichier .bigwig pour UCSC navigateur génome visualisation.
- Télécharger le logiciel 'bedtools de partir du site Web https://github.com/arq5x/bedtools2 et installer le logiciel dans le système 31. Télécharger de bedGraphToBigWig «les outils UCSC du site http://hgdownload.cse.ucsc.edu/admin/exe/ et installer le logiciel dans le système.
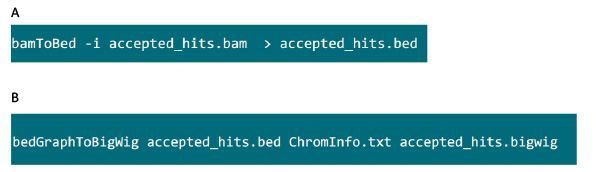
- Dans le dossier contenant le fichier de .bam, utilisez la commande à la figure S6A pour convertir le fichier généré par .bam tophat dans le fichier .bed.
- Une fois le fichier .bed est produit, utilisez la commande à la figure S6B pour générer un fichier .bigwig. Le fichier 'ChromInfo.txt »peut être obtenu à partir de l'URL suivante:Arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Observer un circuit personnalisé sur navigateur UCSC Genome. Consultez le site Web http://genome.ucsc.edu/goldenPath/help/customTrack.html sur la façon d'afficher une piste personnalisée à l'aide UCSC navigateur génome.

Figure S1: Conversion de fichier .bcl à .fastq fichier en utilisant un logiciel de CASAVA.

Figure S2: Cartographie lit pour référencer génome en utilisant Tophat.
Figure S3: détection de nouveaux relevés de notes et estimation du niveau d'expression.

Figure S4: Appel différentiel gène exprimé à l'aide DESeq paquet.

Figure S5: Identification des facteurs de transcription exprimés de manière différentielle.

Figure S6: Conversion résultat de la cartographie pour la visualisation des données.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Afin d'analyser les gènes exprimés de manière différentielle dans Lin-CD34 + et les cellules Lin-CD34 EML, nous avons utilisé la technologie de l'ARN-Seq. La figure 1 montre le flux de travail des procédures. Après isolement de cellules de la lignée négatifs par tri magnétique de cellules, nous nous sommes séparés Lin-SCA + CD34 + et les cellules Lin-SCA-CD34 en utilisant FACS Aria. EML cellules Lin-enrichis ont été colorées avec anti-CD34, anti-Sca1 et anticorps de cocktail de la lignée. Seules les cellules Lin- ont été bloqués pour l'analyse de Sca1 et CD34 expression. Deux populations (SCA + CD34 + et les cellules SCA-CD34- EML) ont pu être observés par l'analyse FACS (figure 2) 6.
Après la séparation des cellules, nous avons extrait l'ARN total à partir de cellules CD34 + et les cellules CD34 respectivement et analysé la qualité de l'ARN. La précision des données d'ARN-Seq repose en grande partie sur la qualité de la bibliothèque d'ARN-Seq et la qualité de l'ARN total est vital pour la préparation d'une bibliothèque de grande qualité. Haute qualité échantillon d'ARN devrait avoir une valeur OD entre 260/280 1.8 et 2,0. En plus d'utiliser le spectrophotomètre, la qualité de l'ARN a été en outre évaluée avec une plus grande précision par Bioanalyseur. La figure 3 montre un résultat d'un échantillon d'ARN de haute qualité avec le RIN égal à 9,4. Seulement haute qualité échantillon d'ARN total avec valeur RIN supérieur à 9 a été utilisé pour l'extraction de l'ARNm et les procédures de construction de bibliothèques suivantes.
ARN ribosomal est le type d'ARN dans la cellule la plus abondante. Actuellement, deux stratégies principales, l'appauvrissement de l'ARNr ou de sélection positive de l'ARNm polyadénylé (poly-A de l'ARNm), sont utilisés pour l'enrichissement de l'ARN cible avant de construction de la banque. Les espèces non d'ARN polyadénylés sont perdus lors de la sélection de l'ARNm poly-A. En revanche, ARNr méthodes d'épuisement tels que RiboMinus pourraient préserver les espèces non-ARN polyadénylés. Le but de notre étude est de chercher expression différentielle des gènes codant dans deux types de cellules, donc nous avons utilisé le poly-A méthode de sélection de l'ARNm de l'enrichissement des ARN cibles avant constru bibliothèquection. Lors de la construction de bibliothèque a été terminée, la taille des fragments d'ADN de la bibliothèque a été vérifiée avant séquençage en utilisant Bioanalyseur. La figure 4 montre une bonne qualité de la bibliothèque avec les pics de la taille de fragment d'environ 300 pb.
Dans l'étape suivante, la banque a été soumis à un séquençage à haut débit. En principe, la durée de lecture plus sera utile pour la cartographie de lecture. Il peut réduire la probabilité que la lecture est associée à plusieurs endroits en raison de la similitude entre les gènes dupliqués ou membres de la famille de gènes. Comme les séquences de séquençage paire de gamme sont les deux extrémités des fragments, la durée de lecture choisie doit être inférieure à la moitié de la longueur moyenne des fragments. Si l'objectif principal de l'expérience est de mesurer le niveau au lieu de construire la structure de transcription d'expression, seule la fin de lecture (75 ou 100 pb) peut réduire le coût sans perdre trop d'informations. Séquençage Jumelé-end est plus utile pour la construction de la structure de transcription et courtlire la longueur peut être utilisé pour réduire les coûts. Certes, lorsque des fonds suffisants sont disponibles, la durée de lecture plus est préférable.
Pour l'analyse de l'expression différentielle, il ya beaucoup d'autres algorithmes autres que DESeq. Il est également l'un inclus dans le forfait de boutons de manchette nommé cuffdiff 32. DESeq est l'un des algorithmes d'analyse de gènes DE LA comptent le plus largement utilisé en fonction. Méthode DESeq est basée sur un modèle statistique bien caractérisé - loi binomiale négative. Dans notre expérience, DESeq est plus stable à comparer cuffdiff. Les premières versions de cuffdiff donnent souvent sensiblement différents nombres de gènes DE. Nous avons donc utilisé pour l'analyse DESeq DE ici.
Parce que les facteurs de transcription jouent un rôle crucial pour la détermination du destin cellulaire, nous nous sommes concentrés sur la transcription significativement exprimés de manière différentielle facteurs de 33. Le TF a changé> 1,5 fois entre Lin-CD34 + et CD34-Lin ont été trouvés et sont indiqués sur la carte de température (Figure 5) 2. En particulier, le taux d'expression relative des TCF7 dans Lin CD34 + des cellules est supérieure à 100 fois supérieure à celle dans les cellules Lin-CD34. Ainsi TCF7 a été choisi pour une ChIP-séquençage (immunoprécipitation de la chromatine et séquençage) analyse et test fonctionnel pour confirmer le fonctionnement de l 'TCF7 dans la régulation de l'EML cellule auto-renouvellement et de différenciation 2.

Figure 1:. Flux de travail des procédures Lin-CD34 + et CD34 Lin-cellules ont été séparées par le système magnétique de séparation de cellules et de cellules activé par fluorescence méthode de tri. L'ARN total a été extrait suivi d'une purification de l'ARNm et de la construction de la bibliothèque. Après analyse de la qualité de la bibliothèque, les échantillons ont été soumis à un séquençage à haut débit. Les données ont été analysées et exprimées de manière différentielle des facteurs de transcription ont été identifiés.

Figure 2: Séparation de Lin-CD34 + et CD34 Lin-EML cellules 6 cellules Lin- EML ont été enrichis par tri cellulaire magnétique.. Cellules Lin- ont été colorées avec un anticorps anti-CD34, anti-anticorps Sca1 et de mélange de la lignée. Cellules Lin- ont été bloqués pour l'expression de CD34 et Sca1. Lin-CD34 + SCA + et populations de cellules Lin-CD34-scalaires EML ont été triés.

Figure 3:. Un représentant de haute qualité échantillon d'ARN total La qualité de l'ARN total a été évalué par Bioanalyzer. L'ARN Nombre intégrité est de 9,4 (FU, fluorescence unités).

Figure 4:. Fragments gamme de taille de la bibliothèque Jumelé-End La distribution de la taille de l'ADN de la banque a été analysée en utilisant Bioanalyseur. La plupart des fragments sont dans la gamme de taille de 250 à 500 pb.

Figure 5:. Différentiellement exprimés les facteurs de transcription (> 1,5 de pliage) entre des cellules Lin-CD34 + et les cellules CD34 Lin-2 Pour chaque type de cellule, deux expériences indépendantes ont été réalisées. Gènes régulés à la hausse sont indiqués comme la couleur rouge et les gènes régulés à la baisse sont indiqués comme couleur verte.
Tableau 1: Tampons et culture cellulaire médiums.
| Logiciel | Usage | Référence | |||
| Bowtie 1.2.7 | Utilisé par Tophat pour la cartographie | [28] | |||
| 1.3.3 Tophat | Cartographie relit à génome de référence | [27] | |||
| Boutons de manchette 1.3.0 | Transcriptions construction et l'estimation du niveau d'expression | [29] | |||
| DESeq 1.16.0 | Analyse de l'expression différentielle | [30] | Bedtools 2.18 | Convertir un fichier en fichier .bam .bed | [31] |
| bedGraphToBigWig | Convertir un fichier .bed à .bigwig fichier | http://genome.ucsc.edu/ |
Tableau 2: Liste des logiciels d'analyse de données.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Transcriptome des mammifères est très complexe 34-38. technologie de l'ARN-Seq joue un rôle de plus en plus important dans les études d'analyse du transcriptome, roman détection des transcriptions et seul nucléotide découverte de variation etc. Il présente de nombreux avantages par rapport aux autres méthodes d'analyse de l'expression des gènes. Comme mentionné dans l'introduction, il surmonte les artefacts d'hybridation de micro-réseau et peut être utilisé pour identifier de nouveaux transcrits de novo. Une limitation de l'ARN-séquençage est relative courte durée de lecture comparant à un séquençage Sanger. Toutefois, avec l'amélioration rapide de la technologie de séquençage, lisez longueur est en constante augmentation. Dans ce papier, nous proposons des méthodes détaillées de l'utilisation de cette technologie pour identifier des régulateurs clés potentiels chez la souris cellule EML auto-renouvellement et de différenciation.
La première étape clé de ce protocole est la culture de cellules EML. Bien EML est une lignée de cellules de précurseur hématopoïétiques et il peut êtrepropagée en grande quantité avec SCF. Les conditions de culture de cellules EML nécessite plus d'attention que les lignées de cellules immortalisées habituels. Les cellules doivent être nourris et des passages à une base régulière avec un fonctionnement doux; sinon les cellules pourraient changer dans leurs propriétés d'auto-renouvellement et de différenciation et subir la mort cellulaire. Comme la première étape après avoir recueilli suffisamment de cellules, nous avons isolé les cellules de la lignée négatifs en utilisant un système de tri cellulaire activé magnétique. Puis nous nous sommes séparés CD34 + et les cellules CD34 en utilisant le tri cellulaire activé par fluorescence. Les cellules sont généralement soumises à des passages EML moins 10 générations avant de l'utiliser pour l'extraction d'ARN et le nombre de cellules CD34 + et CD34 devrait être similaire après la séparation. Si les deux populations varient beaucoup en nombre de cellules, il est conseillé de jeter la culture et re-fondre un autre tube de stock de cellules pour la culture.
Après la séparation de cellules CD34 + et CD34 cellule, l'extraction de l'ARN total a été réalisée, une autre étape importante pour cette mudy. ARN de haute qualité est la base pour la construction d'une bibliothèque de haute qualité, qui promet l'exactitude des données de séquençage. Dans cette étape critique de tout contact avec la RNase doit être évitée. Tous les réactifs doivent être RNase. Il est important de porter des gants à tout moment lors de la manipulation de l'ARN. Haut échantillon d'ARN de qualité a une valeur de DO 260/280 entre 1,8 et 2,0. Lors de la collecte de la phase aqueuse contenant de l'ARN, veillez à ne pas porter toute la phase organique avec l'échantillon d'ARN. Tous les solvants organiques résiduels tels que le phénol ou le chloroforme dans l'ARN se traduirait par une DO260 / 280 valeur inférieure à 1,65. Si le / 280 valeur de DO260 est inférieur à 1,65, précipiter l'ARN de nouveau avec de l'éthanol. Après lavage à l'éthanol à 75%, ne pas trop sécher culot d'ARN. Séchage culot d'ARN complètement affecte la solubilité de l'ARN et conduire à un faible rendement de l'ARN.
La prochaine étape clé pour ce protocole est la préparation bibliothèque. Après extraction de l'ARN total, une étape d'utilisation de la DNase pour éliminer l'ADN contaminé is fortement recommandé, puisque la contamination de l'ADN pourrait entraîner sur la mauvaise estimation de la quantité d'ARN total utilisé. Il est recommandé d'effectuer la procédure en aval immédiatement après purification des ARN, depuis le stockage à long terme après congélation-décongélation et procédure, l'ARN se dégrade à un certain degré. Si les étapes suivantes après purification des ARN ne peuvent pas être effectuées immédiatement, stocker l'ARN -80 ° C. Avant l'ARN total est utilisé pour la purification de l'ARNm et la synthèse d'ADNc, la qualité doit toujours être vérifiée. Seulement ARN de haute qualité peut être utilisé pour la préparation bibliothèque. Utilisation de mauvaise qualité ou de l'ARN dégradé pourrait conduire à "se termine sur-représentation des 3. Avant le séquençage, la qualité bibliothèque a été évaluée pour assurer une efficacité maximale de séquençage.
Dans la partie de l'analyse des données, après avoir effectué une série de boutons de manchette sans un transcriptome de référence, nous avons combiné les nouvelles transcriptions avec transcriptions connues pour former une référence .gtf fichier et lancez Tophat et boutons de manchette pour la deuxième fois.Cette procédure de deux points est recommandée, car cette estimation de fournir FPKM plus précis que de courir une seule fois. Après analyse des données, les gènes exprimés de manière différentielle ont été identifiés. Les expériences peuvent être effectuées en aval de valider la fonction des gènes in vitro et in vivo. Dans notre publication précédente 2, nous avons choisi les facteurs de transcription significativement exprimés de manière différentielle et identifié le site de liaison du génome de ces facteurs en effectuant immunoprécipitation de la chromatine et séquençage (ChIP-Seq). En outre, nous avons appliqué shRNA knockdown dosage pour tester l'effet fonctionnel de TCF7. Nous avons constaté que dans les cellules TCF7 knockdown, les gènes régulés à la hausse ont été les gènes fortement enrichies en cellules CD34, alors que les gènes régulés à la baisse ont été trouvés à être enrichi de façon significative dans des cellules CD34 +. Par conséquent, le profil d'expression génique de cellules knock-down TCF7 décalée vers une partie différenciée state.Overall CD34-, en utilisant la cellule EML comme un système modèlecouplé avec la technologie de séquençage de l'ARN-et analyses fonctionnelles, nous avons identifié et confirmé TCF7 comme un régulateur important de la cellule EML auto-renouvellement et de différenciation.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}