

Summary
RNA-sekvensering og bioinformatikk analyser ble brukt til å identifisere betydelig og forskjellig uttrykt transkripsjonsfaktorer i Lin-CD34 + og Lin-CD34- subpopulasjoner av muse EMLcells. Disse transkripsjonsfaktorer kan spille en viktig rolle i å bestemme veksle mellom selvfornyende Lin-CD34 + og delvis differensiert Lin-CD34- celler.
Abstract
Anvendes hematopoetiske stamceller (HSCs) for klinisk transplantasjon behandling for å gjenoppbygge en pasients hematopoetiske system i mange sykdommer slik som leukemi og lymfom. Belyse mekanismene som kontrollerer HSCs selvfornyelse og differensiering er viktig for anvendelse av HSCs for forskning og kliniske anvendelser. Imidlertid er det ikke mulig å oppnå store mengder av HSCs på grunn av deres manglende evne til å proliferere in vitro. For å overvinne dette hinderet, brukte vi en mus benmarg avledet cellelinje, EML (erytroid, myelogen og lymfatisk) cellelinje, som et modellsystem for denne studien.
RNA-sekvensering (RNA-Seq) har blitt stadig mer brukt til å erstatte microarray for genuttrykk studier. Vi rapporterer her en detaljert metode for å bruke RNA-Seq teknologi for å undersøke potensielle viktige faktorer i regulering av EML celle selvfornyelse og differensiering. Protokollen gitt i denne artikkelen er delt inn i tre deler. Den første part forklarer hvordan kultur EML celler og separat Lin-CD34 + og Lin-CD34- celler. Den andre del av protokollen gir detaljerte prosedyrer for total RNA fremstillingen og den etterfølgende bibliotek konstruksjon for high-throughput-sekvensering. Den siste delen beskriver metoden for RNA-Seq dataanalyse og forklarer hvordan å bruke dataene til å identifisere forskjellig uttrykt transkripsjonsfaktorer mellom Lin-CD34 + og Lin-CD34- celler. De mest markant forskjellig uttrykt transkripsjonsfaktorer ble identifisert til å være de potensielle nøkkel regulatorer kontrollerende EML celle selvfornyelse og differensiering. I diskusjonen delen av dette papiret, vi fremheve de viktigste trinnene for vellykket gjennomføring av dette eksperimentet.
Oppsummert gir dette papiret en metode for å bruke RNA-Seq teknologi for å identifisere potensielle regulatorer av selvfornyelse og differensiering i EML celler. De viktigste faktorene er utsatt for nedstrøms funksjonell analyse in vitro, og jegn vivo.
Introduction
Blodkreft stamceller er sjeldne blodceller som ligger hovedsakelig i voksen benmarg nisje. De er ansvarlig for produksjonen av celler som kreves for å fylle blodet og immunsystemet en. Som en slags stamceller, HSCs er i stand til både selvfornyelse og differensiering. Belyse mekanismene som styrer skjebnen avgjørelse HSCs, mot enten selvfornyelse eller differensiering, vil gi verdifull veiledning om manipulering av HSCs for blodsykdom forskere og klinisk bruk 2. Ett problem som møter forskerne er at HSCs kan opprettholdes og utvides in vitro i svært begrenset grad; det store flertallet av deres avkom er delvis differensiert i kultur 2.
For å identifisere viktige regulatorer som styrer prosessene i selvfornyelse og differensiering på et genom-wide skala, har vi brukt en mus primitive blodkreft stamcellelinje EML som modellsystem. Ther cellelinjen ble avledet fra murin benmarg 3,4. Når fôret med ulike vekstfaktorer, kan EML cellene differensieres til erythroid, myeloid og lymfoide celler in vitro 5. Viktigere, kan denne cellelinjen spres i store mengder i kulturmedium som inneholder stamcellefaktor (SCF), og fremdeles beholde sin multipotentiality. EML celler kan deles inn i subpopulasjoner av selvfornyende Lin-SCA + CD34 + og delvis differensiert Lin-SCA-CD34- celler basert på overflatemarkører CD34 og SCA 6. I likhet med kortsiktig HSCs, SCA + CD34 + celler er i stand til selvfornyelse. Når de ble behandlet med SCF, Lin-SCA + CD34 + celler kan raskt regenerere en blandet befolkning av Lin-SCA + CD34 + og Lin-SCA-CD34- celler og fortsette å spre seg seks. De to populasjoner er like i morfologi og har tilsvarende nivåer av c-kit-mRNA og protein 6. Lin-SCA-CD34--celler er i stand til å spre i medier inneholdende IL-3 i stedet for 3-SCF. Unveiling de sentrale myndigheter i EML celle skjebne beslutning vil gi bedre forståelse av cellulære og molekylære mekanismer tidlig i utviklings overgang under hematopoiesen.
For å undersøke de underliggende molekylære forskjeller mellom selvfornyende Lin-SCA + CD34 + og delvis differensiert Lin-SCA-CD34- celler, brukte vi RNA-Seq å identifisere differensielt uttrykte gener. Spesielt har vi fokus på transkripsjonsfaktorer, som transkripsjonsfaktorer er avgjørende for celle skjebne. RNA-Seq er en nylig utviklet tilnærming som utnytter mulighetene i neste generasjons sekvensering (NGS) teknologi for å profilere og kvantifisere RNA transkribert fra genom 7,8. I korte trekk, er total-RNA poly-A-utvalgt og fragmentert som den innledende template.The RNA-templat blir deretter omdannet til cDNA ved hjelp av revers transkriptase. For å kartlegge full-lengde RNA-transkripter, ved hjelp av intakt, ikke-RNA degradert for å konstruere cDNA bibliotek er viktig. For den purpositur for sekvensering, er spesifikke sekvenser adapter montert på begge endene av cDNA. Deretter, i de fleste tilfeller, blir cDNA-molekyler amplifisert ved PCR og sekvensert i en high-throughput måte.
Etter sekvensering, den resulterende leser kan justeres til et referanse genom og en transkriptom database. Antallet lesninger som kartet til referanse genet blir tellet, og denne informasjonen kan brukes til å estimere genekspresjon nivå. Den leser kan også settes sammen uten de novo en referanse genom, muliggjøre studier av transcriptomes i ikke-modellorganismer 9. RNA-seq teknologi har også blitt anvendt for å detektere spleise isoformene 10-12 nye transkripter, 13 og 14 genfusjoner. I tillegg til påvisning av protein-kodende gener, kan RNA-Seq også brukes til å detektere og analysere nye transkripsjonsnivå av ikke-kodende RNA, slik som lange ikke-kodende RNA 15,16, mikroRNA 17, siRNA etc. 18. På grunn av tHan Nøyaktigheten av denne metode er det blitt benyttet for deteksjon av enkelt nukleotidvariasjoner 19,20.
Før advent av RNA-Seq teknologi, microarray var den viktigste metoden som brukes for å analysere genuttrykk profil. Forhånds utformet prober syntetisert og deretter festet til en fast overflate for å danne en microarray glide 21. mRNA utvinnes og omdannes til cDNA. Under revers transkripsjonsprosessen, blir fluorescerende merkede nukleotider innlemmes i cDNA og cDNA kan bli hybridisert til de microarray lysbilder. Intensiteten av signalet hentet fra et bestemt sted, avhenger av mengden av cDNA-binding til den spesifikke sonde på det punktet 21. Sammenlignet med RNA-Seq teknologi, har microarray flere begrensninger. Først avhengig microarray på pre-eksisterende kunnskap om gen merknader, mens RNA-Seq teknologi er i stand til å oppdage nye transkripsjoner ved relativ høyt nivå bakgrunn, noe som begrenser bruken når gene uttrykk nivået er lavt. Dessuten har den RNA-Seq teknologi mye høyere dynamisk område for påvisning (8000 ganger) 7, mens på grunn av bakgrunn og metning av signaler, er begrenset for både høyt og ydmyk uttrykte gener 7,22 nøyaktigheten av microarray. Til slutt, microarray sonder varierer i sine hybridisering effektivitet, som gjør resultatene mindre pålitelige når man sammenligner relative uttrykk nivåer av ulike transkripsjoner innen én prøve 23. Selv om RNA-Seq har mange fordeler over microarray, er dens dataanalyse kompleks. Dette er en av grunnene til at mange forskere fortsatt microarray benytter i stedet for RNA-Seq. Ulike bioinformatiske verktøy er nødvendig for RNA-Seq databehandling og analyse 24.
Blant flere neste generasjons sekvensering (NGS) plattformer, 454, Illumina, SOLID og Ion Torrent er de mest brukte. 454 var den første kommersielle NGS plattformen. I motsetning til de andre sekvenseringsplattformeneslik som Illumina og SOLID, genererer 454 plattformen lenger lese lengde (gjennomsnittlig 700 basis leser) 25. Lengre leser er bedre for innledende karakterisering av transcriptiome grunn av deres høyere montere effektivitet 25. Den viktigste ulempen av plattformen 454 er den høye kostnaden per megabase av sekvensen. Den Illumina og SOLID plattformer generere lyder med økte tall og korte lengder. Kostnaden per megabase av sekvensen er mye lavere enn 454-plattformen. På grunn av det store antallet av kort leser for Illumina og SOLID plattformer, dataanalyse mye mer beregningskrevende. Prisen på instrument og reagenser for sekvensering for Ion Torrent plattformen er billigere og sekvensering er kortere 25. Imidlertid er feilraten og kostnaden per megabase av sekvens høyere sammenlignet med Illumina og SOLID plattformer. Ulike plattformer har sine egne fordeler og ulemper, og krever ulike metoder for dataanalyse. Den plaTForm bør velges basert på sekvense formål og tilgjengeligheten av finansiering.
I denne artikkelen tar vi Illumina RNA-Seq-plattformen som et eksempel. Vi brukte EML celle som et modellsystem for å undersøke de sentrale myndigheter i EML celle selvfornyelse og differensiering, og ga en detaljert metoder for RNA-Seq bibliotek konstruksjon og dataanalyse for uttrykksnivå beregning og romanen transkripsjon deteksjon. Vi har vist i vårt tidligere publikasjon at RNA-seq studier i modellsystemet EML 2, når kombinert med funksjonstest (f.eks shRNA knockdown) tilveiebringe en kraftig tilnærming i forståelsen av den molekylære mekanismen i de tidlige stadier av hematopoietisk differensiering, og kan tjene som en modell for analyse av celleselvfornyelse og differensiering generelt.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML Cell Kultur og Separering av Lin-CD34 + og Lin-CD34- Cells hjelp Magnetic Cell Sorting System og fluorescensaktivert Cell sorteringsmetode
- Utarbeidelse av babyen hamster nyre (BHK) celledyrkingsmedium for stamcellefaktor samling:
- Kultur BHK-celler i DMEM-medium inneholdende 10% FBS i 25 cm2 kolbe (tabell 1) ved 37 ° C, 5% CO2 i en cellekulturinkubator.
- Når cellene vokse til 80-90% konfluens, vaskes cellene en gang med 10 ml PBS. Tilsett 5 ml av 0,25% trypsin-EDTA-oppløsning til monolaget og cellene inkuberes i 1-5 minutter ved romtemperatur (RT) inntil cellene er frittliggende.
- Pipet løsningen opp og ned forsiktig å bryte opp klumper av celler. Tilsett 5 ml komplett DMEM til kolben for å stoppe trypsin-aktivitet. Samle celler ved sentrifugering ved 200 xg i 5 minutter ved RT.
- Fjern mediumet og cellepelleten suspenderes i 10 ml friskt BHK cellekulturmedium. Overføring 2 ml av cellesuspensjonen fra trinn 1.1.4 til en ny 75 cm2 kolbe og tilsett 48 ml friskt BHK cellekulturmedium til kolben.
- Kultur de BHK-celler i to dager og samle kulturmediet. Passasje mediet gjennom et 0,45 mikrometer filter. Lagre mediet i -20 ° C inntil videre bruk.
- EML cellekultur:
- Kultur EML-celler (i suspensjon) i EML basisk medium inneholdende BHK cellekulturmedium (Tabell 1) ved 37 ° C, 5% CO2 i en cellekulturinkubator.
- Opprettholde EML celler ved lav celletetthet (0,5 til 5 x 10 5 celler / ml) med toppen densitet mindre enn 6 x 10 5 celler / ml. Splitte cellene hver 2-3 dager i forholdet 1: 5. Passage EML celler forsiktig og kast kulturen etter passering i 10 generasjoner.
- Nedbryting av avstamning positive celler:
- Høste EML cellene ved sentrifugering ved 200 xg for 5 min, og cellene vaskes en gang med PBS. Samle cellene ved sentrifugering ved 200 xg i 5 min.
- Suspender cellene med PBS og telle celler med en hemocytometer. Bestemme antistoffkonsentrasjonen i den etterfølgende celleseparasjonstrinn i henhold til antall av cellene (henvises det til instruksjonene som tilbys av leverandøren av cellen isolert system).
- Isoler avstamning negative (karmene) celler ved hjelp avstamning antistoff cocktail (cocktail av biotin-konjugert monoklonale antistoffer CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G / C), 7-4 og Ter-119 ) og en magnetisk aktivert celle sorteringssystem i henhold til produsentens instruksjoner.
- Separering av Lin-CD34 + og Lin-CD34- celler:
- Spinne ned de karmene celler fra trinn 1.3.3 ved 200 xg i 5 min. Cellepelleten suspenderes med PBS og telle celler med en hemocytometer.
- Vask cellene to ganger med FACS-buffer og pelletere cellene ved 200 xgi 5 min.
- Etikett fem 1,5 ml mikrosentrifugerør med henholdsvis tallet 1, 2, 3, 4, 5. Resuspender cellene med 100 ul FACS-buffer per 10 6 10 6 celler (celler pr tube).
- Tilsett 1 mikrogram av Anti-Mouse CD34 FITC antistoff til rør 1 og rør 2 og bland rørene forsiktig.
- Inkuber alle rørene ved 4 ° C i 1 time i mørke.
- Legg 0,25 ug av PE-konjugert anti-Sca1 antistoff og 20 ul av APC-konjugert Lineage Cocktail antistoffer mot røret 1, 0,25 ug av PE-konjugert anti-Sca1 antistoff til røret 3, og 20 ul av APC-konjugerte antistoffer til Lineage Cocktail tube 4.
- Bland alle rørene forsiktig og inkuberes cellene ved 4 ° C i ytterligere 30 min i mørket.
- Tilsett 300 ul FACS-buffer til cellene og spinne ned cellene ved 200 x g i 5 min.
- Vask cellene med 500 ul FACS-buffer til tre ganger.
- Resuspender cellepelleten i 500 ul FACS-buffer.
- Bruk cellene i rørene 2, 3, 4 og 5 for å sette opp kompensasjon. Isoler Lin-SCA + CD34 + og Lin-SCA-CD34- celler i røret en bruker FACS Aria.
2. RNA Forberedelse og Bibliotek Construction for High-throughput sekvensering
- Isolering, kvalitet analyse og kvantifisering av RNA:
- Utdrag total RNA fra Lin-CD34 + og Lin-CD34- celler henholdsvis ved hjelp TRIzol følge produserer 'protokoll.
- Fjern det forurensede DNA ved hjelp av deoksyribonuklease I (DNase I) etter fremstillingen protokoll. Eventuelt lagre RNA ved -80 ° C på dette trinnet for videre bruk.
- Vurdere kvaliteten av total RNA bruker Bioanalyzer i henhold til instruksjonene som tilbys av leverandøren. Bruk RNA prøven med RNA Integrity Number (RIN) lager enn ni.
- Biblioteket Bygg og high-throughput sekvensering:
MERK: Denne protokollen beskriver RNA-Seq hjelp Illumina plattform. Forandre sekvense plattformer, ulike bibliotektilberedningsmetoder nødvendig.- Bruk 0,1-4 ug av total RNA med høy kvalitet per prøve for fremstillingen av bibliotek. Normalt 2 pg av total RNA kan ekstraheres fra 10 5 EML celler.
- Bruk en RNA-sekvenseprøveopparbeidelse system for RNA-rensing og fragmentering, første og andre cDNA syntese, end reparasjon, 3 'ender adenylation, adapter ligation og PCR forsterkning, etter de detaljerte standard prosedyrer fra leverandørens anvisninger.
- Positivt velge PolyA mRNA ved hjelp av oligo-dT magnetiske kuler og fragmentere mRNA.
- Utføre revers transkripsjon ved å bruke tilfeldige primere for å oppnå cDNA og deretter syntetisere den andre tråd av cDNA for å generere dobbeltkjedet cDNA.
- Fjern de 3 'overheng og fylle den 5' overheng av DNA-polymerase. Adenylat 3 'ender for å forhindre ligering av cDNA-fragmenter fra hverandre.
- Legg multiplekse indeksering adaptere til begge ender av dscDNA. Utføre PCR for anriking av DNA-fragmenter.
- Mål A260 / A280 for å innhente informasjon om konsentrasjonen av biblioteket ved hjelp av et spektrofotometer.
- Vurdere biblioteket kvalitet og måle størrelsesområdet av DNA-fragmenter ved hjelp av en Bioanalyzer.
3. Data Analysis
For referanse av programvaren som brukes i denne delen, kan du se (tabell 2).
- Data fil behandling for genetisk analyse:
- Konverter .bcl (base samtale fil) filen til .fastq fila med casava programvare (Illumina, versjon 1.8.2).
- Fyr opp "Terminal" i Linux-systemet. Gå til data mappen som inneholder datafil fra en Illumina HiSeq2000 sekvense maskin. Anta at resultatet mappen er 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /', skrivi kommandoen i figur S1A, og skriv inn data-mappen.
- Installer casava 1.8.2 i Linux-systemet. Anta at outputfolder er "unaligned ', bruker du kommandoen i figur S1B å forberede konfigurasjonsfilen for konvertering. Bruk alternativet --fastq-cluster-count 0 å sikre at bare én .fastq filen er opprettet for hver prøve. Den genererte .fastq filen er i GZ format. Pakke den for genetisk analyse (figur S1B).
- Etter 'unaligned mappen har blitt generert, gå til "unaligned" -mappen (figur S1C).
- Bruk kommandoen i figur S1D å starte konvertering prosessen. The 'j' parameter leverer cpu nummeret som skal brukes.
- Etter at systemet er ferdig konvertering prosessen, gå til resultatet mappen under 'unaligned "-mappen (figur S1E).
- Bruk kommandoen i figur S1F </ Strong> for å dekomprimere .fastq.gz filen inn .fastq fil under hver prøve mappe.
- Konverter .bcl (base samtale fil) filen til .fastq fila med casava programvare (Illumina, versjon 1.8.2).
- Oppdage nye transkripsjoner og behandle uttrykket nivået ved hjelp Tuxedo Suite 26:
- Kartlegge parvise end RNA-Seq leser til muse referansen genomet (UCSC versjon MM9, hentet fra http://cufflinks.cbcb.umd.edu/igenomes.html ) ved hjelp Tophat programvaren (versjon 1.3.3) 27, som bruker den Bowtie lese mapper (versjon 0.12.7) 28. Tophat leveres med "-ingen-romanen-juncs" for å forbedre estimeringsnøyaktigheten uttrykksnivå.
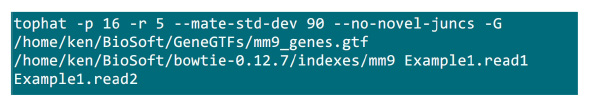
- Sett .fastq filene i en mappe der kartleggingsprosessen vil bli gjennomført. Anta at det er to .fastq filer (endre navn til Example1.read1, Example1.read2) for en sammenkoblet-end sekvense prøve, bruker du kommandoen i Figur S2 å gjøre kartleggingen (justere parametere i henhold til systeminnstilling).Den "-p" parameter leverer cpu nummeret som skal brukes. De "-r" og "-mate-STD-dev" parametere kan fås fra biblioteket QC eller utledes fra en undergruppe av justert leser (figur S2).
- Monter kartlagt leser inn RNA transkripsjoner bruker Mansjettknapper programvaren (versjon 1.3.0) 29. Kjør Mansjettknapper bruker merknadsfil kjente gener (samme .gtf fil som brukes av Tophat) og .bam fil produsert av Tophat.
- Etter Tophat kjørt ferdig, i samme mappe, bruker du kommandoen i figur S3A å kjøre mansjettknapper å konstruere transkriptomet og estimat transkripsjon uttrykk nivå. The 'mm9_repeatMasker.gtf' og genomsekvens filer i 'GenomeSeqMM9' mappen kan fås fra UCSC Genome Browser.
- De resulterende genes.expr og transcripts.expr filer inneholder uttrykket verdien av gener og karakterutskrifter (isoformer). Kopier og lim innfilinnholdet til en Excel-fil og manipulere med regnearkprogram (figur S3b).
- Bruk kommandoen i figur S3C å sammenligne den resulterende 'transcripts.gtf' filen til referanse 'mm9_genes.gtf' fil for å identifisere nye transkripsjoner.
- Den resulterende .tmap filen inneholder sammenligningen resultatet. Kopier og lim filinnholdet til en Excel-fil og manipulere med regnearkprogram. Transkripsjoner med klassekoden 'u' kan betraktes som "roman 'sammenlignet med referanse .gtf filen som følger (figur S3D).
MERK: For genetisk analyse bekvemmelighet sette FPKM verdier til 0,1 hvis verdiene er under 0,1.
MERK: Trinn 3.2.3 - 3.2.6 er valgfritt for de som ønsker å forbedre nøyaktigheten av nye transkripsjoner 'uttrykk estimering. Dette vil ta mye lengre tid, fordi kartlegging og transkriptom konstruksjon må være run mer enn én gang.
- Kjør Tophat bruker standard parametere og deretter kjøre mansjettknapper til generert .gtf fil ved hjelp av kommandoen i Figur S3E.
- Sammenligne den resulterende .gtf filen til referansegenomet .gtf fil ved hjelp av kommandoen i Figur S3F.
- Analysere resulterte .tmap fil som beskrevet i trinn 3.2.2.4. Kopier og lim filinnholdet til en Excel-fil og manipulere med regnearkprogram. Transkripsjoner med klassekoden 'u' kan betraktes som "roman" sammenlignet med referanse .gtf filen gitt.
- Etter trinnet 3.2.5, er det en .combined.gtf fil i mappen som kan anvendes som referanse .gtf fil. En annen kjøring av Tophat og mansjettknapper kan utføres som beskrevet i trinn 3.2.1 og 3.2.2 for å få et mer nøyaktig FPKM estimering av nye transkripsjoner.
- Kartlegge parvise end RNA-Seq leser til muse referansen genomet (UCSC versjon MM9, hentet fra http://cufflinks.cbcb.umd.edu/igenomes.html ) ved hjelp Tophat programvaren (versjon 1.3.3) 27, som bruker den Bowtie lese mapper (versjon 0.12.7) 28. Tophat leveres med "-ingen-romanen-juncs" for å forbedre estimeringsnøyaktigheten uttrykksnivå.
- Oppdage differentially uttrykt gener ved hjelp DESeq pakke 30.
- Input av DESeq er en rå lese teller tabell. For å få et slikt bord, bruke skriptet htseq-count distribueres med HTSeq Python pakke som kan lastes ned fra HTSeq nettside ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Sørg for at samtools, python og htseq-count programsare installert i systemet. Skaff rå lese telle tall fra tophat utgang ved hjelp av kommandoen i Figur S4A.
- Forbered 'Raw_Count_Table.txt', 'ExperimentDesign.txt' filer ved hjelp av Excel. Kopier og lagre innholdet i TXT-format for DESeq R-pakke (figur S4B).
- R installere program i systemet. I terminalen, skriv 'R' og trykk ENTER.A skjermen meldingen vil appearas viste i figur S4C.
- Les 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'i R ved hjelp av kommandoen i Figur S4D.
- Laste DESeq pakken ved hjelp av kommandoen i Figur S4E.
- Faktorforhold i R (figur s4f).
- Bruk kommandoen i figur S4G å kjøre negative binominal test på normalisert telletabell.
- Bruk kommandoen i figur S4H til utgang betydelig differensial uttrykte gener i en CSV-fil.
- Input av DESeq er en rå lese teller tabell. For å få et slikt bord, bruke skriptet htseq-count distribueres med HTSeq Python pakke som kan lastes ned fra HTSeq nettside ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Oppslag transkripsjonsfaktorer '(TFS) FPKM verdier på tvers av prøver ved hjelp av Excel. Snitt DE genet bord og TFS tabellen. Gener tilhører både tabellen er forskjellig uttrykt transkripsjonsfaktorer.
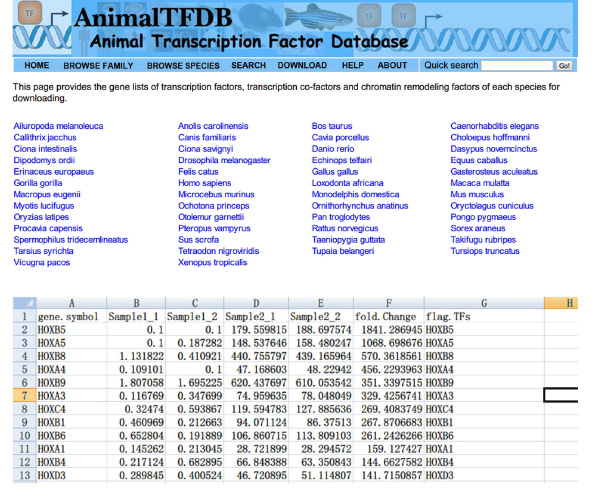
- Gå til nettsiden http://www.bioguo.org/AnimalTFDB/download.php og laste ned transkripsjonsfaktorer. Deretter slå opp DE transkripsjonsfaktorer i Excel (< strong> Figur S5).
- Genererer .bigwig fil for UCSC genom leseren visualisering.
- Last ned 'bedtools' programvarepakke fra nettsiden https://github.com/arq5x/bedtools2 og installere programvaren i systemet 31. Last ned UCSC verktøy "bedGraphToBigWig 'fra nettstedet http://hgdownload.cse.ucsc.edu/admin/exe/ og installere programvaren i systemet.
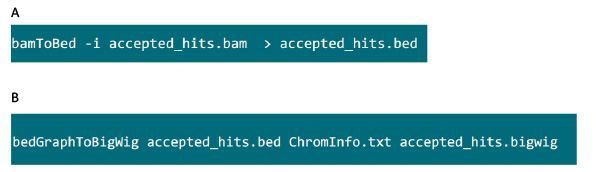
- I mappen som inneholder .bam fil, bruker du kommandoen i figur S6a å konvertere .bam fil generert av tophat inn .bed fil.
- Etter .bed filen er produsert, bruker du kommandoen i figur S6B å generere .bigwig fil. Filen 'ChromInfo.txt' kan fås fra følgende url:Arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Observere en tilpasset spor på UCSC Genome Browser. Gå til nettstedet http://genome.ucsc.edu/goldenPath/help/customTrack.html om hvordan å vise et tilpasset spor ved hjelp av UCSC genom nettleser.

Figur S1: Konvertering .bcl fil til .fastq filen ved hjelp casava programvare.

Figur S2: Mapping leser å referere genomet ved hjelp Tophat.
Figur S3: Påvisning av nye utskrifter og uttrykksnivået estimering.

Figur S4: Calling differensial uttrykt gen ved hjelp DESeq pakken.

Figur S5: Identifisering av forskjellig uttrykt transkripsjonsfaktorer.

Figur S6: Konvertering kartlegging resultat for data visualisering.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
For å analysere differensielt uttrykte gener i Lin-CD34 + og Lin-CD34- EML celler, brukte vi RNA-Seq teknologi. Figur 1 viser arbeidsflyten av prosedyrene. Etter isolering av avstamning negative celler ved magnetisk cellesortering, separert vi Lin-SCA + CD34 + og Lin-SCA-CD34- celler ved hjelp av FACS Aria. Lin-beriket EML cellene ble farget med anti-CD34, anti-Sca1 og avstamning cocktail antistoffer. Bare karmene celler ble gated for analyse av Sca1 og CD34 uttrykk. To populasjoner (SCA + CD34 + og SCA-CD34- EML celler) kunne bli observert av FACS analyse (figur 2) 6.
Etter celleseparasjon, ekstrahert vi total RNA fra CD34 + celler respektivt og CD34- og analysert kvaliteten av RNA. Nøyaktigheten av RNA-Seq data i stor grad avhengig av kvaliteten av RNA-bibliotek Seq og kvaliteten av total RNA er avgjørende for fremstilling av et høykvalitets-bibliotek. Høy kvalitet RNA prøven bør ha en OD 260/280 verdi mellom 1.8 og 2,0. I tillegg til ved hjelp av spektrofotometer, ble RNA kvalitet videre undersøkt med større nøyaktighet ved Bioanalyzer. Figur 3 viser et resultat av en høy kvalitet RNA-prøven med RIN lik 9,4. Høy kvalitet total RNA prøven med RIN verdi som er større enn 9 ble anvendt for mRNA-ekstraksjon og etterfølgende bibliotekkonstruksjonsfremgangsmåter.
Ribosomale RNA er den mest tallrike typer av RNA i cellen. I dag to hovedstrategier, utarming av rRNA eller positivt utvalg av polyadenylert mRNA (poly-A mRNA), brukes til anrikning av target RNA før biblioteket konstruksjon. Ikke polyadenylerte RNA arter går tapt under valg av poly-A mRNA. I motsetning til dette kunne rRNA uttømming metoder som RiboMinus bevare ikke-polyadenylerte RNA-arter. Hensikten med vår studie er å se etter forskjellig uttrykt koding gener i to celletyper, og dermed vi brukte poly-A mRNA valgmetode for anriking av mål RNA før biblioteket construDette skjer. Ved konstruksjon av bibliotek var ferdig, størrelsen av DNA-fragmenter i biblioteket ble kontrollert før sekvensering ved hjelp av Bioanalyzer. Figur 4 viser en god kvalitet bibliotek med de fragmentstørrelse topper ved omtrent 300 bp.
I det etterfølgende trinn, ble biblioteket underkastes high-throughput-sekvensering. I prinsippet vil lenger lese lengde være nyttig for lesekartlegging. Det kan redusere sannsynligheten for at lese er adressert til flere steder på grunn av likheten blant dupliserte gener eller genet familiemedlemmer. Som paret-endesekvense sekvenser er fra begge ender av fragmentene, bør leselengden velges å være mindre enn halvparten av den gjennomsnittlige lengde fragmenter. Hvis det viktigste målet med forsøket er å måle uttrykket nivå i stedet for å konstruere avskrift struktur, lese single-end (75 eller 100 bp) kan redusere kostnadene uten å miste for mye informasjon. Parvise end sekvensering er mer nyttig for transkripsjon struktur konstruksjon og kortereles lengde kan brukes til å redusere kostnadene. Gjerne, når tilstrekkelig kapital tilgjengelig, er lenger lese lengde foretrekkes.
For ekspresjon differensialanalyse, er det mange andre enn DESeq alternative algoritmer. Det er også en inkludert i mansjettknapper pakken heter cuffdiff 32. DESeq er en av de mest brukte count basert DE genet analyse algoritmer. DESeq metoden er basert på en godt karakterisert statistikk modell - negative binomiske fordelingen. Vår erfaring er DESeq mer stabil i forhold til cuffdiff. Tidlige versjoner av cuffdiff ofte gi vesentlig forskjellig antall DE gener. Derfor brukte vi DESeq for DE analysen her.
Fordi transkripsjonsfaktorer er avgjørende for celle skjebne besluttsomhet, fokuserte vi på vesentlig forskjellig uttrykt transkripsjonsfaktorer 33. TFS endret> 1,5 ganger mellom Lin-CD34 + og Lin-CD34- ble funnet og vises på heatmap (Figur5 e) 2. Spesielt, er den relative ekspresjonsnivået av Tcf7 i Lin-CD34 + celler mer enn 100 ganger høyere enn den i Lin-CD34- celler. Dermed Tcf7 ble valgt for videre chip-sekvensering (Kromatin Immunpresipitering og sekvensering) analyse og funksjonstest for å bekrefte Tcf7 's funksjon i regulering av EML celle selvfornyelse og differensiering to.

Figur 1:. Arbeidsflyt av prosedyrene Lin-CD34 + og Lin-CD34- celler ble separert ved magnetisk celleseparasjonssystem og fluorescens-aktivert cellesorteringsmetode. Total RNA ble ekstrahert mRNA etterfulgt av rensing og konstruksjon av bibliotek. Etter analyse av kvaliteten av bibliotek, ble prøver underkastet høy gjennomstrømning sekvensering. Data ble analysert og forskjellig uttrykt transkripsjonsfaktorer Det ble identifisert.

Figur 2: Separering av Lin-CD34 + og Lin-CD34- EML celler seks karmene EML celler ble beriket av magnetisk cellesortering.. Karmene cellene ble farget med anti-CD34, anti-Sca1 og avstamning blanding antistoffer. Karmene celler ble gated for uttrykk av CD34 og Sca1. Lin-CD34 + SCA + og Lin-CD34-SCA- EML cellepopulasjoner ble sortert.

Fig. 3: Et representativt av høy kvalitet total RNA prøven Kvaliteten av total-RNA ble bestemt ved Bioanalyzer. RNA Integrity Number er 9.4 (FU, fluorescens Units).

Figur 4:. Fragmenter størrelse rekke parvise End bibliotek DNA-størrelsesfordeling av biblioteket ble analysert ved anvendelse av Bioanalyzer. De fleste fragmenter er innenfor det størrelsesområde på 250-500 bp.

Figur 5:. Forskjellig uttrykt transkripsjonsfaktorer (> 1,5 fold) mellom Lin-CD34 + celler og Lin-CD34- celler 2 For hver celletype, ble to uavhengige eksperimenter utført. Oppregulert gener er angitt som rød farge og ned-regulerte gener er angitt som grønn farge.
Tabell 1: Buffer og cellekultur medier.
| Programvare | Usage | Referanse | |||
| Bowtie 1.2.7 | Brukes av Tophat for kartlegging | [28] | |||
| Tophat 1.3.3 | Kartlegging leser tilbake til referansegenomet | [27] | |||
| Mansjettknapper 1.3.0 | Transkripsjoner konstruksjon og uttrykk nivå estimering | [29] | |||
| DESeq 1.16.0 | Differential uttrykk analyse | [30] | Bedtools 2.18 | Konverter .bam filen inn .bed fil | [31] |
| bedGraphToBigWig | Konverter .bed fil til .bigwig fil | http://genome.ucsc.edu/ |
Tabell 2: Liste over programvare for dataanalyse.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Pattedyr transkriptomet er svært kompleks 34-38. RNA-Seq teknologi spiller en stadig viktigere rolle i studier av transkriptom analyse, roman transkripsjoner deteksjon og enkelt nukleotidvariasjon funn etc. Det har mange fordeler fremfor andre metoder genekspresjonsanalyser for. Som nevnt i innledningen, overvinner det hybridiseringsreaksjonene gjenstander av microarray og kan brukes til å identifisere nye transkripsjoner de novo. En begrensning av RNA-sekvensering er relativ kort leselengde sammenlignet med Sanger-sekvensering. Imidlertid, med den raske forbedring av sekvenseringsteknologi, les lengde øker stadig. I denne artikkelen gir vi detaljerte metoder for å bruke denne teknologien til å identifisere potensielle nøkkel regulatorer i mus EML celle selvfornyelse og differensiering.
Den første viktig skritt for denne protokollen er EML cellekultur. Selv om EML er en forløper hematopoetisk cellelinje, og den kan blidyrket i store kvantum med SCF. Dyrkingen tilstand EML celler krever mer oppmerksomhet enn de vanlige immortaliserte cellelinjer. Cellene skal fôres og passaged på en jevnlig basis med skånsom drift; ellers cellene kunne endre i sine egenskaper til selvfornyelse og differensiering og gjennomgå celledød. Som det første trinn etter å samle nok celler, har vi isolert avstamning-negative celler ved anvendelse av en magnetisk aktivert cellesorteringssystem. Deretter skilles vi CD34 + og CD34- celler ved hjelp av fluorescens-aktivert celle sortering. EML cellene er normalt passaged mindre enn 10 generasjoner før du bruker for RNA ekstraksjon og antall CD34 + og CD34- celler bør være lik etter separasjon. Hvis de to bestandene varierer sterkt i celle nummer, er det tilrådelig å forkaste kultur og re-tine en annen tube av celle lager for kultur.
Etter separasjon av CD34 + og CD34- celle, ble total RNA ekstraksjon utført, et annet viktig skritt for denne stUdy. Høy kvalitet RNA er basis for konstruksjon av en høykvalitets-bibliotek, som løfter nøyaktigheten av sekvenseringsdata. I dette kritiske trinn, bør enhver kontakt med RNase unngås. Alle reagenser skal RNase gratis. Det er viktig å bruke hansker til enhver tid ved håndtering av RNA. Høy kvalitet RNA prøven har en OD 260/280 verdi mellom 1,8 og 2,0. Ved innsamling av vannfasen som inneholder RNA, være forsiktig med å bære noen organiske fasen med RNA prøven. Eventuelle rester av organiske løsningsmidler slik som fenol eller kloroform i RNA ville resultere i en OD260 / 280 verdi som er lavere enn 1,65. Dersom OD260 / 280 verdien er lavere enn 1,65, RNA utfelles på nytt med etanol. Etter vask med 75% etanol, ikke Overtørkings RNA pellet. Tørking RNA pelleten fullstendig vil påvirke løseligheten av RNA og føre til lavt utbytte av RNA.
Den neste viktige skritt for denne protokollen er biblioteket forberedelse. Etter total RNA ekstraksjon, et trinn med å bruke DNase for fjerning av forurenset DNA jegs sterkt anbefalt, siden DNA-kontaminering kan resultere i feil estimering av mengden av total-RNA brukes. Det anbefales å utføre fremgangsmåten nedstrøms umiddelbart etter RNA isolering, siden etter lang tids lagring og fryse-tineprosedyren, vil RNA nedbrytes til en viss grad. Hvis ikke kan utføres de påfølgende skritt etter RNA isolering umiddelbart, lagre RNA i -80 ° C. Før total RNA blir brukt for mRNA rensing og cDNA-syntese, må kvaliteten alltid kontrolleres. Høy kvalitet RNA kan brukes for fremstillingen av bibliotek. Ved hjelp av lav kvalitet eller degradert RNA kan føre til overrepresentasjon av 3 'slutter. Før sekvensering, ble biblioteket kvalitet vurderes for å sikre maksimal sekvense effektivitet.
I dataanalysen del, etter å ha utført en serie med mansjettknapper uten en referanse transkriptomet kombinerte vi de nye transkripsjoner med kjente transkripsjoner for å danne en referanse .gtf fil og kjøre Tophat og mansjettknapper for andre gang.Denne to-run prosedyre er anbefalt, siden dette gir mer nøyaktig estimering FPKM enn å kjøre én gang. Etter at dataanalyse ble de differensielt uttrykte gener identifisert. Nedstrøms eksperimenter kan utføres for å bekrefte funksjonen av gener in vitro og in vivo. I vår forrige publisering to, valgte vi markant forskjellig uttrykt transkripsjonsfaktorer og identifisert genomet bindingssetet av disse faktorene ved å utføre kromatin immunopresipitering og sekvensering (chip-Seq). I tillegg har vi anvendt shRNA knockdown analyse for å teste den funksjonelle virkningen av Tcf7. Vi har funnet at i Tcf7 knockdown celler, opp-regulerte gener var de gener sterkt anriket på CD34- celler, mens nedregulert gener ble funnet å være vesentlig anriket på CD34 + -celler. Derfor genekspresjonen profilen til Tcf7 knockdown celler forskjøvet mot en delvis differensiert CD34- state.Overall, ved hjelp av EML celle som et modellsystemkombinert med RNA-sekvensering teknologi og funksjonelle analyser, har vi identifisert og bekreftet Tcf7 som en viktig regulator av EML celle selvfornyelse og differensiering.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}