

Summary
Die Bildung von Actomyosin-Bündeln in vitro und die Messung der Myosin-Ensemble-Krafterzeugung mittels optischer Pinzette wird vorgestellt und diskutiert.
Abstract
Myosine sind Motorproteine, die ATP hydrolysieren, um entlang der Spuren des Aktinfilaments (AF) zu treten, und sind essentiell für zelluläre Prozesse wie Motilität und Muskelkontraktion. Um ihre krafterzeugenden Mechanismen zu verstehen, wurde Myosin II sowohl auf Einzelmolekülebene (SM) als auch als Motorteams in vitro mit biophysikalischen Methoden wie optischem Trapping untersucht.
Diese Studien zeigten, dass sich das Verhalten der Myosinkrafterzeugung stark unterscheiden kann, wenn man von der Einzelmolekülebene in einer Drei-Perlen-Anordnung zu Gruppen von Motoren übergeht, die auf einer starren Perlen- oder Deckglasoberfläche in einer Gleitanordnung zusammenarbeiten. Diese Assay-Konstruktionen erlauben es jedoch nicht, die Gruppendynamik von Myosin innerhalb der viskoelastischen Strukturhierarchie zu bewerten, wie dies innerhalb einer Zelle der Fall wäre. Wir haben eine Methode mit optischen Pinzetten entwickelt, um die Mechanik der Krafterzeugung durch Myosin-Ensembles zu untersuchen, die mit mehreren Aktinfilamenten interagieren.
Diese Actomyosin-Bündel erleichtern die Untersuchung in einer hierarchischen und konformen Umgebung, die die motorische Kommunikation und die Ensemblekraftabgabe erfasst. Die anpassbare Natur des Assays ermöglicht es, experimentelle Bedingungen zu ändern, um zu verstehen, wie Modifikationen am Myosin-Ensemble, Aktinfilamentbündel oder der Umgebung zu unterschiedlichen Kraftausgängen führen.
Introduction
Motorproteine sind lebenswichtig und wandeln chemische Energie in mechanische Arbeit um 1,2,3. Myosinmotoren interagieren mit Aktinfilamenten, indem sie Schritte entlang der Filamente ähnlich einer Spur machen, und die Dynamik von Aktin-Myosin-Netzwerken führt unter anderem Muskelkontraktion, Zellmotilität, den kontraktilen Ring während der Zytokinese und die Bewegung der Fracht innerhalb der Zelle durch 3,4,5,6,7,8 . Da Myosine so viele wesentliche Rollen spielen, kann ein Versagen der Funktionalität des Myosin-Aktin-Netzwerks zur Entwicklung von Krankheiten führen, wie z.B. Mutationen in der Myosin-Schwerkette, die eine Herzhyperkontraktilität bei hypertropher Kardiomyopathie (HCM) verursachen9,10,11,12,13,14 . Bei der Muskelkontraktion kooperieren einzelne Myosinmotoren, indem sie als Ensemble zusammenarbeiten, um die erforderliche mechanische Energie bereitzustellen, die das relative Gleiten der AFs 4,15,16,17,18 ausführt. Myosinmotoren bilden Querbrücken zwischen AFs und nutzen Konformationsänderungen aufgrund ihres mechanochemischen Zyklus, um sich kollektiv zum Stachelende der ausgerichteten Filamente 17,18,19,20,21 zu bewegen.
Die Entwicklung quantitativer In-vitro-Motilitätsassays auf SM-Ebene unter Verwendung von Techniken wie optischem Trapping hat es ermöglicht, beispiellose Details über die Funktionsweise einzelner Myosinmotoren zu sammeln, einschließlich der Messung der SM-Krafterzeugung und der Schrittgrößen 22,23,24,25,26,27,28,29,30 . Finer et al. entwickelten den optischen Fallenassay "Drei-Perle" oder "Hantel", um die Krafterzeugungsmechanik einzelner Myosin-II-Motoren23,31 zu untersuchen. Da Muskelmyosin II in Teams arbeitet, um AFs zu kontrahieren, aber auf SM-Ebene nicht prozessiv ist, musste die Ausrichtung des optischen Fangassays vom klassischen motorgebundenen Perlenansatz32 neu angeordnet werden. Um den Hanteltest zu bilden, wurden zwei optische Fallen verwendet, um einen AF über einem Myosinmotor zu halten, der an eine an einem Deckglas befestigte Perle gebunden war, und die Kraftabgabe durch den einzelnen Motor wurde durch Bewegungen des AF innerhalb der Falle23 gemessen.
SM-Kräfte und die Verwendung einer Einzelmotor-/Einzelfilament-Assay-Orientierung geben jedoch kein vollständiges Bild über die Krafterzeugung auf Systemebene, da viele Motorproteine, einschließlich Myosin II, nicht isoliert arbeiten und oft nicht als Summe ihrer Teile funktionieren 15,16,17,32,33,34,35,36 . Komplexere Strukturen, die mehr als einen Motor umfassen, der mit mehr als einem Filament interagiert, sind notwendig, um die Synergie von Myosin- und Aktinfilament-Netzwerken besser zu verstehen15,32. Die Hantel-Assay-Orientierung wurde ausgenutzt, um die Erzeugung kleiner Ensemblekräfte zu untersuchen, indem mehrere Myosine an einer Perle befestigt sind oder ein Myosin-dickes Filament an einer Oberfläche befestigt ist und die Motoren mit dem hängenden AF 4,23,34,37,38,39,40 interagieren können.
Andere kleine Ensemble-Assays umfassen einen In-vitro-Filament-Gleitassay, bei dem Myosinmotoren auf eine Deckglasoberfläche beschichtet werden und eine an einen AF gebundene Perle verwendet wird, um die vom Motorteam erzeugte Kraft zu untersuchen 4,35,36,38,39,40,41,42,43 . In beiden Fällen sind die Myosine an eine starre Oberfläche – Perle oder Deckglas – gebunden und verwenden einen AF. In diesen Fällen sind die Motoren nicht in der Lage, sich frei zu bewegen oder miteinander zu kommunizieren, noch spiegelt die starre Bindung von Myosinen die nachgiebige, hierarchische Umgebung wider, in der die Motoren im Sarkomerzusammenarbeiten würden 32. Frühere Studien haben gezeigt, dass Myosin II seine Umgebung wahrnehmen und sich entsprechend an sich ändernde viskoelastische oder motorische Konzentrationsbedingungen anpassen kann, indem Eigenschaften wie Krafterzeugung und Tastverhältnis41,44,45 verändert werden. Daher besteht die Notwendigkeit, einen optischen Trapping-Assay zu entwickeln, der die motorische Kommunikation und Systemkonformität fördert und erfasst, um ein realistischeres Bild der mechanistischen Grundlagen der Myosin-II-Ensemblekrafterzeugung zu zeichnen.
Hier haben wir eine Methode entwickelt, um hierarchische Strukturen in vitro mit optischem Trapping zu koppeln, indem wir Actomyosin-Bündel oder Sandwiches bilden, die aus mehreren Myosinmotoren bestehen, die zwischen zwei Aktinfilamenten interagieren. Diese modulare Assay-Geometrie hat die Fähigkeit, direkt zu untersuchen, wie molekulare und Umweltfaktoren die Ensemble-Myosin-Krafterzeugung beeinflussen. Darüber hinaus hat die Untersuchung von Krafterzeugungsmechanismen durch diese Aktin-Myosin-Ensembles das Potenzial, bei der Modellierung und dem Verständnis zu helfen, wie sich großräumige zelluläre Aufgaben, wie die Muskelkontraktion, von der molekularen Ebene aus ausbreiten 9,10,13.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Deckgläser ätzen
- 100 g KOH werden in 300 mL 100% Ethanol in einem 1.000 ml Becherglas gelöst. Mit einem Rührbalken umrühren, bis sich der Großteil der KOH aufgelöst hat.
ACHTUNG: Konzentrierte KOH-Lösung kann Verbrennungen und Schäden an der Kleidung verursachen. Tragen Sie Handschuhe, Augenschutz und einen Laborkittel. - Deckgläser einzeln in Deckglas-Reinigungsgestelle legen.
HINWEIS: Die Racks sind mit Schlitzen ausgestattet, die einzelne Deckgläser im Abstand halten, um das Ätzen und Spülen auf jeder Seite des Deckglases zu ermöglichen, Abflusslöcher im Boden und aus Material, das den rauen Ätzbedingungen standhält. Sie können maßgefertigt oder im Handel erworben werden. - Bereiten Sie drei 1.000-ml-Becher vor und kennzeichnen Sie sie: eines mit 300 ml Ethanol und zwei Becher mit 300 ml Umkehrosmosewasser (RO).
HINWEIS: Hier wurde RO-Wasser aus einem Laborwasserreiniger bezogen, könnte aber auch kommerziell erworben werden, wenn kein lokaler Luftreiniger verfügbar ist. - Legen Sie jedes der vier Becher in einen Badebeschaller, um 5 Minuten lang zu entgasen.
- Tauchen Sie ein Gestell mit Deckgläsern in das Becherglas mit KOH und Ethanol und beschallen Sie es für 5 min.
- Übertragen Sie das Deckglasgestell aus dem KOH/Ethanol-Becherglas in das reine Ethanolbecherglas. Tauchen Sie das Gestell im Becherglas auf und ab, bis keine Sicken mehr vorhanden sind.
HINWEIS: Achten Sie darauf, die Deckgläser nicht zu stören oder das Gestell gewaltsam in das Becherglas fallen zu lassen. Dies führt dazu, dass die Deckgläser aus dem Gestell kommen oder chemische Spritzer verursachen. - Übertragen Sie vorsichtig das Gestell mit Deckgläsern aus dem Ethanolbecher in ein Becherglas mit Wasser und tauchen Sie auf und ab, bis keine Abperlen mehr vorhanden sind.
- Tauchen Sie das Gestell mit Deckgläsern in das Becherglas mit noch nicht verwendetem Wasser und beschallen Sie es erneut für 5 Minuten.
- Verwenden Sie eine Flasche, um das Gestell mit Deckgläsern mit Wasser zu besprühen, bis es glatt von den Deckgläsern abläuft. Wiederholen Sie dies mit dem Ethanol.
- Stellen Sie die Racks 20 min in einem Ofen bei 90 °C trocknen. Lagern Sie die Regale mit geätzten Deckgläsern bei Raumtemperatur in geschlossenen Behältern, um eine Kontamination vor dem Gebrauch zu vermeiden.
2. Aktinfilamentpolymerisation
- Lösung T erstellen
- In einem 50 ml konischen Röhrchen 3,94 g Tris-HCl und 0,147 g CaCl2 zugeben. Fügen Sie RO-Wasser hinzu, um ein Gesamtvolumen von 50 ml zu erhalten, und mischen Sie gut.
HINWEIS: Die Endkonzentrationen von Lösung T betragen 500 mM Tris-HCl bzw. 20 mM CaCl2 . - Beschriften Sie das Röhrchen Solution T und lagern Sie es bei 4 °C.
- In einem 50 ml konischen Röhrchen 3,94 g Tris-HCl und 0,147 g CaCl2 zugeben. Fügen Sie RO-Wasser hinzu, um ein Gesamtvolumen von 50 ml zu erhalten, und mischen Sie gut.
- TC Buffer erstellen
- Mischen Sie 40 mL RO-Wasser und 1,5 ml Lösung T in einem 50-ml-konischen Röhrchen. Ändern Sie den pH-Wert auf 8,0, indem Sie kleine Mengen konzentrierten KOH hinzufügen. Fügen Sie Wasser hinzu, um 50 ml der Lösung herzustellen, und überprüfen Sie den pH-Wert. Stellen Sie bei Bedarf den pH-Wert ein.
HINWEIS: Der endgültige TC-Puffer enthält 5 mM Tris-HCl und 0,2 mMCaCl2 bei pH 8. - Beschriften Sie das Röhrchen TC und lagern Sie es bei 4 °C.
- Mischen Sie 40 mL RO-Wasser und 1,5 ml Lösung T in einem 50-ml-konischen Röhrchen. Ändern Sie den pH-Wert auf 8,0, indem Sie kleine Mengen konzentrierten KOH hinzufügen. Fügen Sie Wasser hinzu, um 50 ml der Lösung herzustellen, und überprüfen Sie den pH-Wert. Stellen Sie bei Bedarf den pH-Wert ein.
- FC-Puffer erstellen
- 85 ml RO-Wasser, 10 ml Lösung T, 3,73 g KCl und 0,041 g MgCl2 in eine 100-ml-Pufferflasche geben. Modifizieren Sie den pH-Wert auf 7,5, indem Sie kleine Mengen konzentrierten KOH hinzufügen. Fügen Sie Wasser hinzu, um ein Endvolumen von 100 ml zu erhalten, und überprüfen Sie den pH-Wert.
HINWEIS: Der endgültige FC-Puffer enthält 500 mM Tris-HCl, 500 mM KCl, 2 mM MgCl 2 und 2 mM CaCl2 bei pH 7,5. - Beschriften Sie die Tube FC und lagern Sie sie bei 4 °C.
- 85 ml RO-Wasser, 10 ml Lösung T, 3,73 g KCl und 0,041 g MgCl2 in eine 100-ml-Pufferflasche geben. Modifizieren Sie den pH-Wert auf 7,5, indem Sie kleine Mengen konzentrierten KOH hinzufügen. Fügen Sie Wasser hinzu, um ein Endvolumen von 100 ml zu erhalten, und überprüfen Sie den pH-Wert.
- Bereiten Sie General Actin Buffer (GAB) vor.
- Mischen Sie 485 μL TC-Puffer, 10 μL 10 mM ATP und 5 μL 50 mM DTT in einem Mikrozentrifugenröhrchen.
HINWEIS: Die endgültigen Pufferbedingungen sind 5 mM Tris-HCl, 0,2 mM CaCl 2, 0,5 mM DTT und0,2 mM ATP. - Beschriften Sie es als GAB und lagern Sie es bei 4 °C.
- Mischen Sie 485 μL TC-Puffer, 10 μL 10 mM ATP und 5 μL 50 mM DTT in einem Mikrozentrifugenröhrchen.
- Aktinpolymerisationspuffer (APB) vorbereiten.
- Mischen Sie 455 μL FC-Puffer, 25 μL 100 mM ATP und 20 μL 50 mM DTT in einem Mikrozentrifugenröhrchen.
HINWEIS: Die endgültigen Pufferbedingungen sind 50 mM Tris-HCl, 500 mM KCl, 2 mM MgCl 2, 2 mM CaCl 22 mM DTT und 5 mM ATP. - Beschriften Sie das Röhrchen als APB und lagern Sie es bei 4 °C.
- Mischen Sie 455 μL FC-Puffer, 25 μL 100 mM ATP und 20 μL 50 mM DTT in einem Mikrozentrifugenröhrchen.
- Aktin rekonstituieren
- Rekonstituieren Sie Kaninchen-Skelettmuskel-Aktin durch Zugabe von 100 μL deionisiertem Wasser zu einer 1-mg-Durchstechflasche mit lyophilisiertem Aktin. Gut mischen, indem Sie vorsichtig auf und ab pipettieren. Aliquot in 5 μL Proben, einfrieren und die 10 mg/ml Aktin-Aliquots bei -80 °C lagern.
- Rekonstituieren Sie biotinyliertes Kaninchen-Skelettmuskel-Aktin durch Zugabe von 20 μL RO-Wasser. Aliquot in 5 μL Proben, snap-freeze und Lagerung der 1 mg/ml biotinylierten Aktin-Aliquots bei -80 °C.
- Unmarkierte Aktinpolymerisation mit Rhodaminphalloidin-Stabilisierung
- Eine Durchstechflasche mit 10 mg/ml Aktin auftauen und auf Eis halten.
- Bereiten Sie frischen GAB-Puffer vor, geben Sie 100 μL GAB zum Aktin-Aliquot und mischen Sie durch vorsichtiges Pipettieren auf und ab. Die Lösung 1 h auf Eis inkubieren.
- Bereiten Sie frisches APB während der Inkubation zu. Nach der Inkubation polymerisieren Sie das Aktin zu Filamenten, indem Sie der Aktinlösung 11 μL APB hinzufügen. Gut mischen, indem Sie vorsichtig auf und ab pipettieren. 20 min auf Eis legen.
- Geben Sie 5 μL rhodaminmarkiertes Phalloidin in die frisch polymerisierte Aktinfilamentlösung. 1 h im Dunkeln auf Eis lassen.
- Lagern Sie die in Aluminiumfolie eingewickelte Durchstechflasche mit Rhodaminaktin im Dunkeln bei 4 °C.
HINWEIS: Es wird empfohlen, diese Filamente für einen Zeitraum von maximal 1 Woche zu verwenden. Die AF-Qualität kann jeden Tag durch eine schnelle Bildgebung einer Flusszelle, die nur AFs enthält, und die tägliche Betrachtung konsistenter Filamente bestätigt werden.
- Biotinylierte Aktinpolymerisation mit Alexa Fluor 488 Phalloidin-Stabilisierung
- Eine Durchstechflasche mit 10 mg/ml Aktin und 1 Durchstechflasche mit 1 mg/ml biotinyliertem Aktin auftauen und auf Eis halten.
- Erstellen Sie einen frischen GAB-Puffer.
- Kombinieren Sie die beiden Durchstechflaschen (Schritt 2.8.1) in einem Aktin:Biotinyl-Aktin-Verhältnis von 10:1. Geben Sie 100 μL GAB zu der Aktinmischung und mischen Sie gut, indem Sie vorsichtig auf und ab pipettieren. Auf Eis 1 h inkubieren.
- Machen Sie frisches APB während der Inkubation.
- Nach dem Inkubationsschritt polymerisieren Sie das Aktin durch Zugabe von 11 μL APB zur Aktinlösung. Gut mischen, indem Sie vorsichtig auf und ab pipettieren. Auf Eis 20 min inkubieren.
- Fügen Sie 5 μL Alexa Fluor 488-markiertes Phalloidin hinzu und inkubieren Sie 1 h auf Eis im Dunkeln.
- Bewahren Sie die in Aluminiumfolie eingewickelte Biotin-Durchstechflasche im Dunkeln bei 4 °C auf.
HINWEIS: Diese Filamente können für einen Zeitraum von maximal 1 Woche verwendet werden.
3. Myosin und Perlenzubereitung
- Rekonstituieren Sie Myosin II
- Drehen Sie kurz (~ 5 s) lyophilisiertes Skelettmyosin II herunter, um es am Boden der Röhre mit einer Standard-Minizentrifuge zu sammeln.
- Rekonstituieren Sie das Myosin zu 10 mg/ml durch Zugabe von 100 μL 1 mM DTT, hergestellt in RO-Wasser.
- Die Stamm-Myosinlösung wird 10x verdünnt, indem man 10 μL 10 mg/ml Myosin zu 90 μL 1 mM DTT in RO-Wasser hinzufügt. Aliquots mit kleinem Volumen (1-5 μL) herstellen, einfrieren und bei -80 °C lagern.
HINWEIS: Die Myosinaktivität kann durch Durchführung eines Standard-Gleitfilament-Assays bestätigt werden, wie zuvorveröffentlicht 46,47. Eine kurze Beschreibung finden Sie in der Diskussion.
- Reinigung von Streptavidin-beschichteten Perlen
- 20 μL 1 μm Streptavidinperlen in 80 μL RO-Wasser verdünnen. Viermal waschen, indem man es bei 9.600 × g herunterdreht und in 100 μL RO-Wasser rekonstituiert.
- Für 2 min bei 40% Amplitude beschallen und die gewaschenen Perlen auf einem Rotator bei 4 °C lagern.
4. Vorbereitung der Durchflusszelle
- Bereiten Sie eine Poly-L-Lysin-Lösung (PLL) vor, indem Sie 30 ml 100% Ethanol zu einem 50-ml-Röhrchen geben und 200 μL 0,1% w/v Poly-L-Lysin in Wasser hinzufügen und gut mischen.
- Ein geätztes Deckglas in die PLL-Lösung geben und 15 min einweichen lassen. Entfernen Sie das Deckglas mit einer Pinzette, und achten Sie darauf, nur den Rand des Deckglases zu berühren, wenn es aus dem Schlauch gezogen wird (siehe Abbildung 1A-C). Greifen Sie die Deckgläser mit einer behandschuhten Hand an den Kanten.
- Trocknen Sie das Deckglas mit einer gefilterten Fluggesellschaft, bis kein Ethanol mehr vorhanden ist und keine Rückstände mehr auf dem Deckglas vorhanden sind.
- Tragen Sie zwei Stücke doppelseitiges Klebeband auf die Mitte eines Objektträgers auf, die 3-4 mm voneinander entfernt sind. Reißen oder schneiden Sie das überschüssige Klebeband ab, das am Rand der Folie hängt.
- Fügen Sie das PLL-beschichtete Deckglas auf dem Band senkrecht zur Längsachse des Objektträgers hinzu (ein T bildend), um einen Kanal zu bilden.
- Verwenden Sie einen kleinen Tubus, um das Deckglas gründlich auf das Band und den Objektträger zu komprimieren, bis das Band transparent ist (Abbildung 1A). Stellen Sie sicher, dass sich keine Blasen im Band befinden, da dies zu Leckagen aus dem Durchflusskanal führen kann.
HINWEIS: Die Durchflusszelle kann ein Volumen von 10-15 μL aufnehmen.
5. Actomyosin-Bündel-Vorbereitung
- In separaten Röhrchen wird jede Art von Aktinfilament (rhodamin- und biotinyliert 488-markiert) 600x verdünnt, indem 0,5 μL des jeweiligen, markierten Aktins mit 300 μL APB gemischt werden. Zusätzlich 5 μL des entsprechend markierten Phalloidins in jedes Röhrchen geben und 15 min auf Eis im Dunkeln inkubieren.
- Zu der biotinylierten Aktinlösung wird ein Sauerstofffängersystem aus 1 μL Beta-D-Glucose bei 500 mg/ml, 1 μL Glucoseoxidase bei 25 mg/ml und 1 μL Katalase bei 500 Einheiten/ml hinzugefügt. Fügen Sie 1 μL 100 mM ATP und 1 μL 100x verdünnte, gereinigte Streptavidin-Perlen hinzu. Vorsichtig mit einer Pipettenspitze umrühren. Setzen Sie die Suspension auf einen Rotator bei 4 °C, während der Rest des Actomyosin-Bündels zusammengebaut wird.
- Geben Sie 15 μL des verdünnten Rhodamin-Aktins in die PLL-Durchflusszelle (Abbildung 1D). Leiten Sie die überschüssige Lösung durch die Durchflusszelle, aber lassen Sie den Strömungskanal nicht trocken werden. 10 min in einer Feuchtigkeitskammer inkubieren.
HINWEIS: Feuchtigkeitskammern können aus leeren Pipettenspitzenkästen hergestellt werden, in denen Wasser auf den Boden gegeben wird und der Deckel mit Aluminiumfolie bedeckt ist, um Licht zu blockieren. - Bereiten Sie eine 1 mg/ml Caseinlösung in APB vor.
- Fügen Sie 15 μL 1 mg/ml Casein hinzu, um eine unspezifische Bindung der nachfolgenden Komponenten zu verhindern (Abbildung 1E). 5 min in einer Feuchtigkeitskammer inkubieren.
- Geben Sie die gewünschte Konzentration von Myosin zu der biotinylierten Aktin- und Perlensuspension aus Schritt 5.2. Rühren Sie vorsichtig mit der Pipettenspitze um und geben Sie dann sofort 15 μL der Stufe 5.2 Suspension + die gewünschte Myosinkonzentration in die Flusszelle (Abbildung 1F, G). Inkubieren für 20 min. Versiegeln Sie die offenen Enden der Durchflusszelle mit Nagellack, um Verdunstung während bildgebender und optischer Fangexperimente zu verhindern.
HINWEIS: Eine Myosinlösungskonzentration von 1 μM ergibt eine robuste Bündelung und kann als Ausgangspunkt für die gewünschte Anpassung des Assays verwendet werden (siehe Abbildung 2).
6. Kraftmessungen mit optischer Falle (NT2 Nanotracker2)
HINWEIS: Während das folgende Protokoll speziell für das NT2-System gilt, kann dieser Assay mit anderen optischen Fangsystemen verwendet werden, einschließlich solcher, die speziell angefertigt sind und ebenfalls über Fluoreszenzfähigkeiten verfügen. Der allgemeine Arbeitsablauf bleibt derselbe, die Oberfläche des Objektträgers scharf zu stellen, Perlenkalibrierungen durchzuführen und Daten zu erfassen, indem fluoreszierende Aktinbündel gefunden werden. Für das NT2-System enthalten die ergänzende Abbildung S1, die ergänzende Abbildung S2, die ergänzende Abbildung S3, die ergänzende Abbildung S4, die ergänzende Abbildung S5, die ergänzende Abbildung S6 und die ergänzende Abbildung S7 Einzelheiten zum optischen Fangsystem und zur Softwareschnittstelle.
- Schalten Sie die Steuerbox und den Laser ein (Zusatzfigur S1).
- Starten Sie die optische Falle-Computersoftware, indem Sie auf das JPK Nanotracker-Symbol auf dem Desktop klicken.
- Wecken Sie die Fernbedienung auf, indem Sie auf die Logitech-Taste in der Mitte klicken (Zusatzabbildung S2).
- Schalten Sie das Fluoreszenzmodul ein, indem Sie den Ein-/Ausschalter umschalten (Ergänzende Abbildung S3).
- Drehen Sie den Filterwürfelturm für die Hellfeld-Bildgebung (ergänzende Abbildung S4).
- Sobald das System fertig ist, schalten Sie den Laser mit der Laser-Power-Taste in der linken unteren Ecke des Bildschirms auf 50 mW ein und lassen Sie ihn 30 Minuten lang stabilisieren (ergänzende Abbildung S5).
- Klicken Sie nacheinander auf die Schaltflächen Beleuchtung, Kamera, Objektiv und Bühnenbewegung in der Software, um diese Fenster zur Anzeige und Manipulation während des Experiments aufzurufen. Schalten Sie die Mikroskopbeleuchtung ein, indem Sie auf die Schaltfläche On/Off klicken und sie auf maximale Leistung einstellen, indem Sie klicken und den Balken ganz nach rechts ziehen (Ergänzende Abbildung S5).
- Öffnen Sie den Probenbereich und nehmen Sie den Probenhalter vom Mikroskoptisch. Fügen Sie die Durchflusszelle hinzu, sichern Sie sie mit den Metallprobenhaltern und stellen Sie sicher, dass sich der Objektträger mit dem Deckglas auf der Unterseite befindet.
- Fügen Sie 30 μL RO-Wasser in die Mitte des unteren Objektivs hinzu. Lassen Sie die Pipettenspitze die Linse nicht berühren. Setzen Sie den Probentisch wieder ein.
HINWEIS: Da das NT2-System ein Wasserimmersionsobjektiv als Fangobjektiv verwendet, können die Tauchmedien je nach Fangobjektiv in der Konfiguration des Benutzers unterschiedlich sein. - Heben Sie das untere Objektiv mit den Kontrollpfeilen auf dem Bildschirm oder L2 auf der Fernbedienung an, bis die Wasserperle das Deckglas berührt (ergänzende Abbildung S5).
- Senken Sie das obere Objektiv mit den Bildschirmpfeilen oder R2 auf der Fernbedienung ab, bis etwa die Hälfte der Entfernung zur Durchflusszelle erreicht ist. Geben Sie 170 μL RO-Wasser in die Oberseite der Durchflusszelle direkt unter dem oberen Objektiv. Senken Sie das obere Objektiv, bis es die Oberflächenspannung des Wassers bricht und einen Meniskus bildet.
- Bewegen Sie den Mikroskoptisch mit dem Pfeilpad auf der Fernbedienung, bis der Rand des Bandes neben dem Strömungskanal erreicht ist. Schließen Sie die Probenklappe.
HINWEIS: Ein "Klick" beim Schließen der Probenklappe zeigt an, dass der Laserverschluss nun geöffnet ist. Dies ist eine Sicherheitsfunktion, die das Öffnen des Rollladens nur bei geschlossener Tür zulässt. - Bringen Sie im Objektivfenster auf dem Bildschirm den Rand des Bandes scharf, indem Sie das untere Objektiv mit dem Namen Laserobjektiv nach oben bringen, indem Sie mithilfe der Bildschirmsteuerelemente auf den oberen Pfeil klicken. Machen Sie dasselbe für das oberste Ziel, indem Sie auf den unteren Pfeil klicken (ergänzende Abbildung S5).
HINWEIS: Die Doppelpfeile verschieben das Ziel oder die Stufe schneller. Der Rand des Bandes wird zum Fokussieren verwendet, da es sich um ein großes, leicht zu findendes Objekt handelt, das sich in der Nähe der Deckglasoberfläche befindet. Luftblasen innerhalb des Bandes sind eine weitere Option. Dies ist jedoch nicht erforderlich, wenn der Benutzer über eine automatisierte Routine zur Ermittlung des Oberflächenfokus oder eine bevorzugte interne Methode verfügt. - Sobald das Band scharf ist, schließen Sie die Iris an der Oberseite der optischen Falle teilweise. Bringen Sie das obere Objektiv nach unten, bis die Polygonform der Iris sichtbar ist. Bringen Sie diese Kanten in den Fokus, öffnen Sie die Blende wieder und koppeln Sie dann die Ziele mit, indem Sie auf das Vorhängeschloss-Symbol klicken (Ergänzende Abbildung S5).
- Finden Sie eine schwimmende Perle und fangen Sie sie ein, indem Sie auf den Trap Shutter Button klicken, der den Verschluss öffnet und es dem Trapping-Laser ermöglicht, die Probe zu treffen. Klicken Sie auf den Trap-Cursor auf dem Bildschirm und ziehen Sie ihn, um die Position des Trapping-Lasers zu verschieben. Nach dem Einfangen kalibrieren Sie die Perle, um Spannungsmessungen mit Kraft und Verschiebung zu korrelieren.
- Klicken Sie auf die Schaltfläche Kalibrierung . Passen Sie die Kalibrierungsroutine basierend auf der Leistungsspektrenanalyse an und passen Sie die Eckfrequenz innerhalb der Software für die X-, Y- und Z-Richtung an (ergänzende Abbildung S6).
- Klicken Sie auf Einstellungen. Geben Sie den Durchmesser der Perle (1.000 nm) und die Temperatur der Stufe unten links im Softwarefenster ein. (siehe ergänzende Abbildung S6).
- Klicken Sie auf Trap 1. Klicken Sie auf X-Signal. Klicken Sie auf Ausführen , um die Eckfrequenzanpassung durchzuführen. Klicken und ziehen Sie innerhalb des Fensters, um die Funktionsanpassung zu optimieren. Klicken Sie auf Verwenden Sie es für Empfindlichkeits- und Steifigkeitswerte. Klicken Sie auf Werte akzeptieren. Wiederholen Sie diesen Vorgang für die Y- und Z-Signale. Schließen Sie das Fenster. (siehe ergänzende Abbildung S6).
HINWEIS: Perlenkalibrierungsroutinen auf anderen optischen Fangsystemen oder kundenspezifischen Systemen, die vom Benutzer robust getestet wurden, wie z. B. die Gleichverteilungsmethode oder die Widerstandskraftmethode, sind ebenfalls akzeptabel57,58. - Finden Sie ein Actomyosin-Bündel, indem Sie nach Perlen suchen, die an AFs auf der Oberfläche des Deckglases gebunden sind.
- Wenn eine Perle erkannt wird, die nicht von anderen schwimmenden Perlen überfüllt ist, beobachten Sie die AFs um sie herum durch Fluoreszenzbildgebung, um das Vorhandensein eines Bündels zu überprüfen.
- Stellen Sie sicher, dass ein Bündel vorhanden ist, indem Sie nach beiden fluoreszierenden AFs suchen, die kolokalisiert sind. Schalten Sie die Weißlichtquelle ein und verwenden Sie den entsprechenden Filterwürfel, um jedes Aktinfilament abzubilden, indem Sie den Turm drehen (488 nm bzw. 532 nm Anregungsfilterwürfel für Alexa Fluor 488 bzw. Rhodaminanregung). Siehe ergänzende Abbildung S4.
HINWEIS: Ein Kontrollexperiment zur Überprüfung der Fluoreszenzintensität einzelner AFs kann nützlich sein, um Bündel zu identifizieren, die aus einem einzigen 488- und einem einzelnen Rhodamin-markierten Filament bestehen, oder anwendbar auf den Satz von Fluorophoren, den der Benutzer verwendet. - Sobald Sie dies überprüft haben, fangen Sie die Perle ein, die am oberen Filament des Bündels befestigt ist, indem Sie auf den Trap Shutter-Button klicken.
- Verwenden Sie die Bildschirmsteuerung, um die Daten aufzuzeichnen, indem Sie auf die Oszilloskop-Schaltfläche klicken (ergänzende Abbildung S7). Um Messungen zu visualisieren, ohne die Daten aufzuzeichnen, klicken Sie auf Start. Um alle Daten zu speichern, klicken Sie auf Automatisch speichern. Um Messungen aufzuzeichnen, klicken Sie auf Aufzeichnung starten. Wählen Sie, welche Daten in Echtzeit visualisiert werden sollen (Position, Kraft, x-Richtung, y-Richtung), indem Sie aus dem Dropdown-Menü X-Signal oder Y-Signal auswählen. Denken Sie daran, dass xdirection von links nach rechts und y-Richtung auf dem Bildschirm oben und unten ist. Siehe ergänzende Abbildung S7.
HINWEIS: Die Daten werden als .out-Dateien gespeichert und enthalten Zeit, Spannung, Verschiebung und Kraft für jede Richtung. Diese Dateien können zur Visualisierung und Analyse in andere Software exportiert werden.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Die Durchflusszellen, die die Actomyosin-Bündelsysteme enthalten, haben ein Standarddesign, bestehend aus einem Objektträger und einem geätzten Deckglas, das durch einen Kanal aus doppelseitigem Klebeband getrennt ist (Abbildung 1). Der Assay wird dann vom Deckglas bis zur Verwendung von abgestuften Einführungen, wie im Protokoll beschrieben, erstellt. Der endgültige Assay besteht aus Rhodamin-markierten Aktinfilamenten; die gewünschte Myosinkonzentration (1 μM wurde für die repräsentativen Ergebnisse in Abbildung 2 und Abbildung 3 verwendet); biotinylierte, Alexa Fluor 488-markierte Aktinfilamente; 1 μm Streptavidin-Perlen; das Sauerstofffängersystem; ATP; und APB-Puffer. Pro Durchflusszelle werden mehrere Bündel gebildet, und die oben beschriebenen Aktinkonzentrationen ergeben einen ausreichenden Abstand zwischen den Bündeln, um sicherzustellen, dass keine unerwünschten Wechselwirkungen auftreten. Dies erleichtert auch die Durchführung mehrerer Kraftmessungen pro Durchflusszelle, um die Effizienz der Datenerfassung zu erhöhen. Kraftprofile sollten innerhalb einer Durchflusszelle und von Flusszelle zu Durchflusszelle reproduzierbar sein.
Während das obige Protokoll auf die Verwendung eines kommerziellen optischen Fangaufbaus ausgerichtet ist, könnten die hier vorgestellte Flusszelle und der Assay leicht für ein anderes kommerzielles Instrument oder einen speziell angefertigten optischen Fangaufbau in Verbindung mit einem Mikroskop oder Mikroskoptisch verwendet werden und über Fluoreszenzbildgebungsfunktionen verfügen. Sobald alle Flusszellenzugaben gemäß dem obigen Protokoll abgeschlossen sind, sind die Actomyosin-Bündel auf dem Objektträger (Abbildung 1) sofort messbereit. Die Durchflusszelle wird dem optischen Fallenmikroskoptisch hinzugefügt, mehrere Perlenkalibrierungsmessungen werden erfasst und Bündel werden durch Fluoreszenzkolokalisierung der Bündelfilamente identifiziert. Eine an ein Bündel gebundene Perle wird gefangen, und die Verschiebungs- und entsprechende Kraftmessung beginnt. Der Benutzer kann die Datenerfassung in Echtzeit auf dem Computermonitor beobachten. Abhängig von der Konzentration von Myosin, das in der Flusszelle verwendet wird, kann das Bündel sofort eine erhebliche Bewegung zeigen, oder es kann 30 s-1 min dauern, um effektiv eine Zunahme der Verschiebung / Kraft zu sehen.
Eine repräsentative Kraftspur ist in Abbildung 3A dargestellt, wo die Myosinmotoren eine stetige Kraftrampe aufweisen, gefolgt von einem Plateau. Es ist typisch zu sehen, wie sich diese Art von Spuren über 2-5 Minuten entwickeln. Es ist aber auch möglich, Actomyosin-Bündel zu messen, die keine Nettokraft erzeugen (Abbildung 3B). Diese Spuren erscheinen als Grundlinienrauschen oder weisen keinen wesentlichen Nettokraftanstieg über 90 s auf. Dies ist wahrscheinlich auf eine geringe lokale Konzentration des Motors zurückzuführen, die kein produktives Gleiten zulässt, oder das Bündel befindet sich in einer ungünstigen parallelen Ausrichtung, in der die Plus- und Minusenden der Filamente ausgerichtet sind.
Da der Inhalt der Durchflusszelle anfällig für eine Verschlechterung durch den einfallenden Beleuchtungs- und Einfanglaser, die lokale Erwärmung des Objektträgers im Laufe der Zeit und die Erzeugung radikaler Sauerstoffspezies sein kann, wird dringend empfohlen, dieselbe Flusszelle nicht länger als 1 h zu verwenden. Für maximale Effizienz wird empfohlen, einen weiteren Assay während der Datenerfassung inkubieren zu lassen. Die Weg-/Kraftspur kann aus der optischen Überfüllungssoftware zur weiteren Filterung und Analyse in Excel, Matlab, Igor oder andere Datenverwaltungsprogramme exportiert werden. Daten, die aus solchen optischen Traping-Ensemble-/Bündelexperimenten extrahiert werden können, umfassen verschiedene Arten von Krafterzeugungsprofilen (Baseline, Rampe/Plateau) unter variierenden Assay-Bedingungen, Geschwindigkeit der Krafterzeugung, maximale Krafterzeugung, Ensemblekinetik und Schrittverhalten durch Schrittgrößen und Verweilzeiten zwischen Schritten oder Teams von Schritten sowie Tastverhältnis. Der Benutzer kann auch die Assay-Bedingungen ändern, um zu vergleichen, wie die Zugabe verschiedener Arten von Myosinmotoren, die Zugabe von Aktin-bindenden Proteinen oder die Änderung der Pufferbedingungen diese Eigenschaften der Ensemblekrafterzeugung beeinflussen.
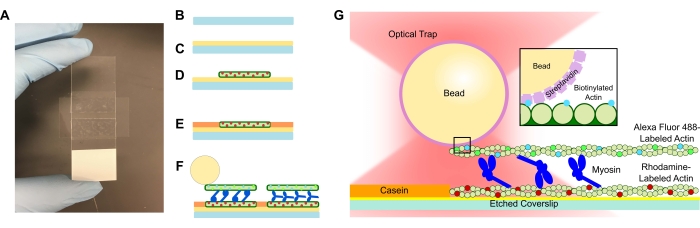
Abbildung 1: Schematische Darstellung des Assays. (A-C) Geätzte Deckgläser werden mit Poly-L-Lysin beschichtet und zur Bildung der Durchflusszelle mit doppelseitigem Klebeband und einem Objektträger verwendet. Zeitgesteuerte Einführungen und Inkubationsschritte, die im Protokoll beschrieben sind, führen zu rhodaminmarkiertem Phalloidin-stabilisiertem Aktin als Schablone oder unteres Filament (D), gefolgt von Kaseinblockierung, um eine unspezifische Bindung zu verhindern (E), und (F) Alexa Fluor 488 Phalloidin-stabilisiertes biotinyliertes Aktin als Fracht- oder Oberfilament und Teams von Myosin II, die die Filamente auseinander schieben und Kraft erzeugen, wenn ATP eingeführt wird. Die Geometrie der Motoren und die Art der Vernetzung innerhalb des Bündels können unter verschiedenen Bedingungen, wie z. B. der Salzkonzentration59, variieren. Frühere Studien haben gezeigt, dass die Myosin-Schwanzdomäne die Fähigkeit hat, mit Aktinfilamenten und langsamer Ensemblemotilität zu interagieren46. Myosinköpfe in schweren Meromyosin-Experimenten zeigen jedoch die Bindung jedes Kopfes an benachbarte Aktinfilamente60. (G) Streptavidin-Perlen werden als optischer Griff für die Falle verwendet und binden sich ausschließlich an das biotinylierte Aktinfilament der Fracht, was zur Validierung beiträgt, dass auf dem Objektträger geeignete Bündel gebildet werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.

Abbildung 2: Fluoreszierende Actomyosin-Bündel. Vier verschiedene Begegnungen von Aktinfilamenten und -bündeln innerhalb des in Abbildung 1 dargestellten Bündelassays. Das obere biotinylierte Aktinfilament mit dem Alexa Fluor 488 Phalloidinkanal ist links und das untere Template-Aktinfilament mit dem Rhodamin-Phalloidinkanal rechts dargestellt. Unten ist die gleiche Figur mit farbigen Linien dargestellt, die überlagert sind, um das Auge zu führen. (A) Ein oberes Aktinfilament befindet sich in der Nähe eines unteren Aktinfilaments, hat aber eine unvollständige Überlagerung. Dies würde nicht für Bündelexperimente verwendet werden. (B) Obere und untere Aktinfilamente sind kolokalisiert, und die Intensität jedes Filaments bestätigt, dass es sich jeweils um einzelne Filamente innerhalb des Bündels handelt. Dies wäre ein guter Kandidat für Bündelexperimente. (C) Ein großes Bündel selbstorganisierter Rhodaminfilamente befindet sich auf der Unterseite. Während es ein entsprechendes oberes Aktinfilament gibt, das kolokalisiert ist, sind zu viele untere Filamente vorhanden; Daher würde es nicht für Bündelexperimente verwendet werden. Dies ist auch ein Beispiel dafür, wie die Fluoreszenzintensität zunimmt, wenn mehrere Aktinfilamente desselben Typs gebündelt werden. Der Benutzer kann dies als Messgerät für die Beurteilung einzelner Filamente im Vergleich zu Bündeln desselben Filamenttyps verwenden. (D) Ein unteres Filament ist ohne entsprechendes oberes Filament vorhanden, was auch bestätigt, dass kein Durchbluten vorhanden ist. Dies würde nicht für Bündelexperimente verwendet werden. Wir stellen fest, dass die Intensität der Filamente im Alexa Fluor 488-Kanal gering ist und glauben, dass dies auf das verwendete Filterset zurückzuführen ist (Filterset 09 von Zeiss). Das für den Rhodaminkanal verwendete Filterset ist das Filterset 43 von Zeiss. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.

Abbildung 3: Myosin II Ensemble Force Generation. Repräsentative Spuren von skelettalen Myosin-II-Motoren, die Kraft innerhalb der konstruierten in vitro Aktinstrukturhierarchie erzeugen. Die Myosinmotoren arbeiten zusammen, um kollektiv und produktiv Kraft zu erzeugen, bis ein Plateau erreicht ist und die Kraft aufrechterhalten wird (A) oder eine Antagonisierung in der Nähe der Basislinie (B) erfährt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
Ergänzende Abbildung S1: Bruker/JPK Nanotracker2 optische Falle. (A) Computermonitor. (B) Computertastatur. (C) Computerturm. (D) Controller-Box. (E) Laserstromversorgung. (F) Optischer Fallenoptikkasten. (G) Inverses Mikroskop. (H) Tür zum Mikroskoptisch. (I) Polarisator-Schieberegler zum Umschalten zwischen Hellfeld- und differentieller Interferenzkontrastbildgebung. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S2: Fernbedienung für optische Falle. (A) Tastatur zur Positionierung der motorisierten Bühne. (B-C) Stellen Sie die Trap-Position ein. (D) A, X und B schalten den Hauptverschluss ein und aus, fallen 1 Verschluss bzw. Trap 2 Verschluss. (E) Die Logitech-Taste wird verwendet, um den Controller aufzuwecken. (F) Die Aufwärts- und Abwärtstasten, die zum Positionieren des Fangobjektivs verwendet werden. (G) Die Aufwärts- und Abwärtstasten, die zur Positionierung des Erkennungsobjektivs verwendet werden. Beachten Sie, dass die Fernbedienung nicht erforderlich ist und alle diese Manipulationen in der Software durchgeführt werden können. Es ist jedoch praktisch, die Objektive und die Tischposition steuern zu können, während Sie in die Umgebung des Mikroskoptisches schauen. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S3: Fluoreszenzmodul für optische Falle. Die 89North PhotoFluor Fluoreszenz-Weißlichtquelle ist an die Rückseite des inversen Mikroskops gekoppelt. Es wird mit einem Kippschalter (Pfeil) ein- und ausgeschaltet. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S4: Fluoreszenzfilter-Würfelrevolver. Der Revolver (Pfeil) kann gedreht werden, um den Filterwürfel zu verwenden, der für die Bildgebung in DIC-, Rhodamin- oder Alexa Fluor 488-Farbstoffen erforderlich ist. Beachten Sie, dass Filterwürfel ausgetauscht werden können, um das Setup für die Verwendung verschiedener Fluorophore anzupassen. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S5: Nanotracker2 Software. (A) Laser-Netzschalter und Steuerung. (B) Objektives Positionierungsfenster. Richtungspfeile werden verwendet, um die Erkennungs- (oben) und Überfüllungsziele (unten) zu verschieben. Doppelpfeile bewegen die Objektive mit einer höheren Geschwindigkeit. Der blau-rote Knopf unten links entkoppelt die Objektive und zieht sie wieder in ihre ursprüngliche Position zurück. Dies ist notwendig, wenn Proben in und aus dem Mikroskoptisch entnommen werden. Die dritte Taste von links mit dem Objektiv- und Vorhängeschloss-Symbol "koppelt" die Objektive, so dass der Benutzer sowohl die Fang- als auch die Erkennungsobjektive in der z-Achse nach oben und unten bewegen kann, wenn sie beide scharf sind und Kohler-Beleuchtung erreichen. (C) Probenpositionierungsfenster zum Bewegen des Mikroskoptisches in der x- und y-Achse. Doppelpfeile bewegen die Bühne mit einer höheren Geschwindigkeit. Dieses Fenster wird durch Klicken auf das Symbol nach oben/unten und links / rechts im oberen Menü aktiviert. (D) Kamera-Visualisierungsfenster. Das Schraubenschlüsselsymbol kann verwendet werden, um benutzerdefinierte Bildbedingungen festzulegen. Dieses Fenster wird durch Klicken auf das Kamerasymbol im oberen Menü aktiviert. (E) Mikroskopbeleuchtungsfenster. Dieses Fenster wird durch Klicken auf das Glühbirnensymbol im oberen Menü aktiviert. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S6: Kalibrierfenster. (A) Dieses Fenster dient der Perlenkalibrierung und wird durch Klicken auf das Cal-Symbol im oberen Menü aktiviert. Um eine Perle zu kalibrieren, wird eine optimale Anpassung der Eckfrequenz in den x-, y- und z-Signalen erreicht. (B) Wählen Sie für jedes Signal die entsprechende Signaltaste oben links. (C) Klicken Sie auf Ausführen und optimieren Sie die Passform, indem Sie innerhalb des grünen Fensters (D) klicken und ziehen. (E) Wenn Sie mit der Passform zufrieden sind, klicken Sie auf Verwenden Sie es für Empfindlichkeit und Steifigkeit. Dies ermöglicht die Erfassung der Verschiebung in Nanometern und der Kraft in Pikonewton. (F) Klicken Sie dann unten links auf Werte akzeptieren . Wiederholen Sie diesen Vorgang für die y- und z-Richtungen. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S7: Fenster Datenerfassung. Dieses Fenster dient zur Erfassung von Positions- und Kraftdaten und ermöglicht es dem Benutzer, die Messungen in Echtzeit zu sehen. (A) Dieses Fenster wird durch Klicken auf das x,t-Symbol im oberen Menü aktiviert. (B) Der Benutzer kann zwischen der Anzeige der x- und y-Signale wechseln. (C) Klicken Sie auf Start , um mit der Visualisierung der Daten zu beginnen. Klicken Sie auf Autosave, um die Daten zu speichern. Klicken Sie auf Aufnahme starten , um mit der Aufzeichnung und Speicherung der Daten zu beginnen. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Eine In-vitro-Studie mit optischen Pinzetten in Kombination mit Fluoreszenzbildgebung wurde durchgeführt, um die Dynamik von Myosin-Ensembles zu untersuchen, die mit Aktinfilamenten interagieren. Aktin-Myosin-Aktin-Bündel wurden unter Verwendung von Muskelmyosin II, Rhodamin-Aktin an der Unterseite des Bündels und auf der Deckglasoberfläche und 488-markierten, biotinylierten Aktinfilamenten auf der Oberseite des Bündels zusammengesetzt. Aktinprotein aus Kaninchenmuskeln wurde unter Verwendung von allgemeinen Aktinpuffern (GAB) und Aktinpolymerisationspuffern (APB) polymerisiert und stabilisiert. GAB und APB müssen täglich frisch im Labor mit ATP, FC-Puffer und TC-Puffer zubereitet werden. Muskelmyosin II wurde verwendet, um die Aktin-Myosin-Aktin-Sandwiches zu bilden. Phalloidin wurde zur Fluoreszenzfärbung der Aktinfilamente sowie zur Stabilisierung in vitro verwendet.
Die Myosinaktivität kann durch Durchführung eines Standard-Gleitfilament-Assays bestätigt werden, wie zuvor veröffentlicht46,47. Myosin II und seine Subfragmente können in einer Vielzahl von Orientierungen an die Deckglasoberfläche binden, und das Vorhandensein der Schwanzdomäne kann das Gleiten des Filaments im Vergleich zu Assays mit schwerem Meromyosin46,48,49 verlangsamen. Gleiten und Oberflächenbewegungen können jedoch weiterhin beobachtet werden. Eine offensichtlichere Demonstration der Myosinaktivität ist das aktive Brechen von Aktinfilamenten, das beobachtet werden kann, wenn längere Aktinfilamente in kleinere Fragmente zerlegt werden, die dann in mehrere Richtungen weggleiten. Dies geschieht aufgrund der hohen Konzentration aktiver Motoren auf der Oberfläche, wurde von mehreren Laboratorien beobachtet und tritt nicht ohne aktive Myosinmotoren 42,50,51,52,53,54 auf. Darüber hinaus hilft der hier vorgestellte Bündelassay bei der Linderung von Motilitätsproblemen, die in erster Linie mit dem Gleitfilament-Assay in Verbindung gebracht wurden, wie z.B. die Vielfalt der motorischen Bindungsorientierungen auf einem Glasdeckglas, da der Bündelassay eine Kaseinblockierung der Glasoberfläche beinhaltet, so dass Motoren innerhalb des Bündels 47,55,56 binden.
Der erste Schritt besteht darin, Rhodamin-Aktin-Filamente als Boden- oder Schablonenfilament zu einem Poly-L-Lysin-beschichteten Deckglas in einer Durchflusszelle hinzuzufügen. Poly-L-Lysin wird verwendet, um die Aktinbindung zu fördern, da Polylysin positiv geladen ist, während Aktin negative Ladungen aufweist und in früheren Zytoskelett-In-vitro-Assay-Präparaten 61,62,63 verwendet wurde. Vor der Bündelbildung wurden einer Flusszelle verschiedene Aktinverdünnungen zugesetzt, um die Aktinkonzentration zu optimieren. In diesem Fall war 600x aus dem Bestand die optimale Verdünnung, die eine ausreichende Anzahl von Schablonenfilamenten für die Bündelbildung ergab, jedoch mit ausreichendem Abstand, so dass Bündel individualisiert wurden. Die Verdünnung erfolgte mit dem APB-Puffer. Nach der Zugabe von Rhodamin-Aktin folgte eine Schicht Kasein, um die Oberfläche zu blockieren und eine unspezifische Bindung zu vermeiden. Die Durchflusszelle wurde für 30 min inkubiert und nach der Inkubation mit Puffer gewaschen, um ungebundene Aktinfilamente auszuwaschen. Schließlich wurde der Flusszelle eine Kombination aus Myosin, 488 / Biotin-Aktin und Streptavidin-beschichteten Perlen hinzugefügt, um die Bildung von Aktin-Myosin-Bündeln zu erleichtern. Die Kugelkonzentration sollte so sein, dass genügend vorhanden sind, um oberflächengebundene Bündel zu binden, und ausreichend in Suspension, um die Kalibrierung zu erleichtern. Eine zu hohe Perlenkonzentration kann jedoch zu Schwierigkeiten bei Fangexperimenten führen, da benachbarte Perlen in die Laserfalle fallen und die Messung stören. Myosinmotoren werden der Kombination unmittelbar vor der Injektion in den Objektträger zugesetzt, so dass die Myosinmotoren nicht präventiv mit dem Ladungs- oder oberen biotinylierten Aktinfilament aggregieren und somit das untere Rhodamin zu biotinylierten Aktinfilamenten binden.
Das optische Fangsystem NT2 ist eine kommerzielle optische Falle mit kombinierten Hellfeld-, Differenzinterferenzkontrast- (DIC) und Epifluoreszenz-Bildgebungsmodalitäten. Es ist mit einem Zeiss AxioObserver 3 inversen Mikroskop mit 100x/NA 1.46 und 63x/NA 1.0 Wassertauchfang- und Detektionsobjektiven gekoppelt. Das System ist mit der Klick- und Schleppfangfunktion einer Laserfalle ausgestattet und kann während der Bildgebung in jeder der aufgeführten Modalitäten verwendet werden. Die gebildeten Bündel werden mittels Fluoreszenzbildgebung detektiert und bestätigt. Eine Weißlichtquelle mit entsprechenden Filterwürfeln (GFP/FITC und TRITC/CY3) ermöglicht einen schnellen Wechsel zwischen der Filamentbildgebung. Kolokalisierte AFs wurden verifiziert, indem die AFs bei den verschiedenen Anregungswellenlängen visualisiert wurden, bevor jede Kraftmessung mit optischen Pinzetten durchgeführt wurde. Da die Filamente auch mit einem Sauerstofffängerreagenz schnell photobleichen können, wird empfohlen, dass Forscher Visualisierungsparameter wie Intensität und Belichtungszeit optimieren, bevor sie die Bündelexperimente durchführen.
Für die Kraftmessungen wurde optisches Trapping verwendet, wobei die Streptavidinperlen in Gegenwart von ATP verwendet wurden, um das biotinylierte Frachtaktinfilament zu binden und die Myosin-Krafterzeugung als Kraftwandler zu aktivieren. Verschiebungs- und Kraft-Zeit-Daten, die durch optisches Trapping gewonnen wurden, wurden zur Analyse aus der Trapping-Software extrahiert. Die kommerzielle Trapping-Software bietet jedoch auch Analyseroutinen, die verwendet werden können, oder benutzerdefinierte Algorithmen in anderen Programmen können vom Benutzer programmiert werden, um Fangdaten zu visualisieren und zu analysieren. Bei kundenspezifischen optischen Fangsystemen kann der Benutzer Anregungslaser anstelle einer Weißlichtquelle mit Filtern verwenden, die ebenfalls akzeptabel sind. Darüber hinaus können Fluoreszenzfarbstoffe geändert werden, um sie an die vorhandene Ausrüstung anzupassen, die ein Benutzer möglicherweise hat, wenn sich die Emissionsspektren nicht überlappen und Durchblutungen verursachen.
Wir stellen fest, dass der vorgestellte Assay ein Basisassay ist, der vom Benutzer abhängig von seiner Forschungsfrage im Bereich der Actomyosin-Ensemblemechanik weiter angepasst werden kann. Der allgemeine Arbeitsablauf kann auch auf andere In-vitro-Zytoskelett-Ensemblesysteme angewendet werden, die von Interesse sein könnten, wie z.B. Mikrotubuli-Bündelassays, die minimale Modelle der mitotischen Spindel 32,61,63,64,65,66 bilden. Zu den Änderungen gehören unter anderem die Änderung der Fluorophor-Markierungen, die für das bestehende Setup des Benutzers geeignet sind. Veränderung der Myosinkonzentration, des Konstrukts oder des Isotyps; und Titrierungspufferbedingungen, unter anderem.
Potenzielle Herausforderungen sind bei der Durchführung dieses Assays möglich. Bei der Bildung der Aktin-Myosin-Bündel ist die Myosinkonzentration innerhalb der Aktinbündel möglicherweise nicht homogen über den Objektträger. Um dies zu berücksichtigen, werden mehrere Bündel über den gesamten Schlitten gemessen, um sicherzustellen, dass Motorverteilungs- und Krafterzeugungsprofile ordnungsgemäß abgetastet werden. Es ist auch schwierig, die Bündelorientierung zu kennen, wenn dies für die Interpretation von Kraftdaten erforderlich ist. Daher sollten für jedes Bündel mehrere Versuche durchgeführt werden. Man könnte auch Aktin-Filament-Endmarkierung durch fluoreszierendes Gelsolin oder Gelsolin-beschichtete Perlen von einer kleineren Größe als der optische Fanggriff einbauen. Die Fluoreszenzbildgebung kann auch verwendet werden, um x- und y-Komponentenkräfte zu betrachten, um die Bündelorientierung abzuleiten. Da der Myosin-Aggregationszustand stark von der Ionenstärke des Puffers beeinflusst wird, wobei bei schneller Verdünnung von KCl dicke Filamente gebildet werden, sollte die Puffersalzkonzentration angemessen überwachtwerden 67,68.
Frühere Studien, die andere In-vitro-Methoden wie Gleitassays verwendeten, waren hilfreich, um die Rolle von Myosindomänen zu identifizieren und die Konfiguration und Wechselwirkungen zwischen Myosin und anderen Aktin-bindenden Proteinen zu untersuchen. Diese Methoden haben jedoch den Nachteil, dass die Bindung von Myosin an eine starre Oberfläche das Koordinationspotenzial zwischen Myosinmotoren und damit die mechanosensorische Rückkopplung begrenzt, die auftritt, um zu bestimmen, ob sich das Motorensemble in einem Modus mit hohem oder niedrigem Tastverhältnis befindet 33,35,41,69. Darüber hinaus gibt das optische Fallen mit Single-Myosin-Motornetzwerken kein klares Verständnis dafür, wie Myosinmotoren miteinander und mit Aktinfilamenten interagieren. Das hier entwickelte Protokoll ermöglicht die Untersuchung der myosinmotorischen Ensembledynamik innerhalb eines konformen, hierarchischen Aktinnetzwerks. Es ist auch in Bezug auf Motor-Filament-Ensemble-Eigenschaften wie Konzentration, Isoform und Pufferumgebung anpassbar, um eine systematische Untersuchung zu ermöglichen. Das vorgestellte Protokoll ist eine Plattform für zukünftige Studien komplexerer Actomyosin-Netzwerke und behält die Präzision von Verschiebungs- und Krafterzeugungsmessungen bei, die durch optisches Trapping erleichtert werden, das traditionell für Einzelmolekülstudien verwendet wurde.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren haben keine Interessenkonflikte zu erklären.
Acknowledgments
Diese Arbeit wird teilweise vom University of Mississippi Graduate Student Council Research Fellowship (OA), dem University of Mississippi Sally McDonnell-Barksdale Honors College (JCW, JER), dem Mississippi Space Grant Consortium unter der Fördernummer NNX15AH78H (JCW, DNR) und der American Heart Association unter der Fördernummer 848586 (DNR) unterstützt.
Materials
| Name | Company | Catalog Number | Comments |
| Actin protein (biotin): skeletal muscle | Cytoskeleton | AB07-A | Biotinylated actin protein |
| Actin protein, rabbit skeletal muscle | Cytoskeleton | AKL99-A | Actin protein |
| Alexa Fluor 488 Phalloidin | Invitrogen | A12379 | Actin stabilizer and Alexa Fluor 488 stain |
| ATP | Fisher scientific | BP413-25 | Required for actin assembly and myosin motility |
| Beta-D-glucose | Fisher scientific | MP218069110 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Blotting Grade Blocker (casein) | Biorad | 1706404 | Used to block surface from non-specific binding |
| CaCl2 | Fisher scientific | C79500 | Calcium chloride, provides the necessary control over the dynamics of actin myosin network |
| Catalase | Fisher scientific | ICN10040280 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Coverslips | Fisher scientific | 12544C | Used to make flow cells |
| DTT | Fisher scientific | AC327190010 | Used for buffer preparation |
| Ethanol | Fisher scientific | A4094 | Regent used for cleaning coverslips |
| Glucose oxidase | Fisher scientific | 34-538-610KU | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| KCl | Fisher scientific | P217-500 | Used for buffer preparation |
| KOH | Fisher scientific | P250-1 | Used to etch coverslips and adjust buffer pH |
| MgCl2 | Fisher scientific | M33-500 | Used for buffer preparation |
| Microscope slides | Fisher scientific | 12-544-2 | Used to make flow cells |
| Myosin II protein: rabbit skeletal muscle | Cytoskeleton | MY02 | Full length myosin motor protein isolated from rabbit skeletal muscle |
| Nanotracker2 | Bruker/JPK | NT2 | Optical trapping instrument |
| Poly-l-lysine | Sigma-Aldrich | P8920 | Facilities adhesion of actin filaments onto glass surface of the coverslip |
| Rhodamine Phalloidin | Cytoskeleton | PHDR1 | Actin stabilizer and rhodamine fluorescent stain |
| Streptavidin beads, 1 μm | Spherotech | SVP-10-5 | Optical trapping handle |
| Tris-HCl | Fisher scientific | PR H5121 | Used for buffer preparation |
References
- Goldstein, L. S. Kinesin molecular motors: transport pathways, receptors, and human disease. Proceedings of the National Academy of Sciences of the United States of America. 98 (13), 6999-7003 (2001).
- Lee Sweeney, H., Holzbaur, E. L. F.
- O'Connell, C. B., Tyska, M. J., Mooseker, M. S. Myosin at work: Motor adaptations for a variety of cellular functions. Biochimica et Biophysica Acta - Molecular Cell Research. 1773 (5), 615-630 (2007).
- Kaya, M., Tani, Y., Washio, T., Hisada, T., Higuchi, H. Coordinated force generation of skeletal myosins in myofilaments through motor coupling. Nature Communications. 8, 1-13 (2017).
- Akhshi, T. K., Wernike, D., Piekny, A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton. 71 (1), 1-23 (2014).
- Brawley, C. M., Rock, R. S. Unconventional myosin traffic in cells reveals a selective actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America. 106 (24), 9685-9690 (2009).
- Hartman, M. A., Spudich, J. A. The myosin superfamily at a glance. Journal of Cell Science. 125 (7), 1627-1632 (2012).
- Spudich, J. A., et al.
- Sommese, R. F., et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proceedings of the National Academy of Sciences of the United States of America. 110 (31), 12607-12612 (2013).
- Nag, S., et al. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nature Structural & Molecular Biology. 24 (6), 525-533 (2017).
- Kawana, M., Sarkar, S. S., Sutton, S., Ruppel, K. M., Spudich, J. A. Biophysical properties of human b-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Science Advances. 3 (2), 1-11 (2017).
- Girolami, F., et al. Novel α-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: A massively parallel sequencing study. Circulation: Cardiovascular Genetics. 7 (6), 741-750 (2014).
- Debold, E. P., et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. American Journal of Physiology - Heart and Circulatory Physiology. 293 (1), 284-291 (2007).
- Barron, J. T.
- Duke, T. A. J.
- Vilfan, A., Duke, T.
- Huxley, A. F. Muscle structure and theories of contraction. Progress in Biophysics and Biophysical Chemistry. 7, 255-318 (1957).
- Huxley, H. E. Fifty years of muscle and the sliding filament hypothesis. European Journal of Biochemistry. 271 (8), 1403-1415 (2004).
- Kad, N. M., Kim, S., Warshaw, D. M., VanBuren, P., Baker, J. E. Single-myosin crossbridge interactions with actin filaments regulated by troponin-tropomyosin. Proceedings of the National Academy of Sciences of the United States of America. 102 (47), 16990-16995 (2005).
- Veigel, C., Molloy, J. E., Schmitz, S., Kendrick-Jones, J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nature Cell Biology. 5 (11), 980-986 (2003).
- Spudich, J. A.
- Simmons, R. M., Finer, J. T., Chu, S., Spudich, J. A. Quantitative measurements of force and displacement using an optical trap. Biophysical Journal. 70 (4), 1813-1822 (1996).
- Finer, J. T., Simmons, R. M., Spudich, J. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 368 (6467), 113-119 (1994).
- Kron, S. J., Uyeda, T. Q. P., Warrick, H. M., Spudich, J. A. An approach to reconstituting motility of single myosin molecules. Journal of Cell Science. 98, 129-133 (1991).
- Molloy, J. E., Burns, J. E., Kendrick-Jones, B., Tregear, R. T., White, D. C. S. Movement and force produced by a single myosin head. Nature. 378 (6553), 209-212 (1995).
- Ruegg, C., et al. Molecular motors: Force and movement generated by single Myosin II molecules. Physiology. 17 (5), 213-218 (2002).
- Nayak, A., et al. Single-molecule analysis reveals that regulatory light chains fine-tune skeletal myosin II function. Journal of Biological Chemistry. 295 (20), 7046-7059 (2020).
- Dupuis, D. E., Guilford, W. H., Wu, J., Warshaw, D. M.
- Tyska, M. J., et al. Two heads of myosin are better than one for generating force and motion. Proceedings of the National Academy of Sciences of the United States of America. 96 (8), 4402-4407 (1999).
- Tyska, M. J., Warshaw, D. M.
- Finer, J. T., et al.
- Al Azzam, O., Trussell, C. L., Reinemann, D. N. Measuring force generation within reconstituted microtubule bundle assemblies using optical tweezers. Cytoskeleton. 78 (3), 111-125 (2021).
- Wagoner, J. A., Dill, K. A. Evolution of mechanical cooperativity among myosin II motors. Proceedings of the National Academy of Sciences of the United States of America. 118 (20), 2101871118 (2021).
- Walcott, S., Warshaw, D. M., Debold, E. P. Mechanical coupling between myosin molecules causes differences between ensemble and single-molecule measurements. Biophysical Journal. 103 (3), 501-510 (2012).
- Stewart, T. J., Murthy, V., Dugan, S. P., Baker, J. E. Velocity of myosin-based actin sliding depends on attachment and detachment kinetics and reaches a maximum when myosin-binding sites on actin saturate. Journal of Biological Chemistry. 297 (5), 101178 (2021).
- Hilbert, L., Cumarasamy, S., Zitouni, N. B., Mackey, M. C., Lauzon, A. M. The kinetics of mechanically coupled myosins exhibit group size-dependent regimes. Biophysical Journal. 105 (6), 1466-1474 (2013).
- Debold, E. P., Walcott, S., Woodward, M., Turner, M. A. Direct observation of phosphate inhibiting the Force-generating capacity of a miniensemble of myosin molecules. Biophysical Journal. 105 (10), 2374-2384 (2013).
- Kaya, M., Higuchi, H. Nonlinear elasticity and an 8-nm working stroke of single myosin molecules in myofilaments. Science. 329 (5992), 686-689 (2010).
- Pertici, I., et al. A myosin II nanomachine mimicking the striated muscle. Nature Communications. 9 (1), 1-10 (2018).
- Cheng, Y. S., De Souza Leite, F., Rassier, D. E. The load dependence and the force-velocity relation in intact myosin filaments from skeletal and smooth muscles. American Journal of Physiology - Cell Physiology. 318 (1), 103-110 (2020).
- Stam, S., Alberts, J., Gardel, M. L., Munro, E. Isoforms confer characteristic force generation and mechanosensation by myosin II filaments. Biophysical Journal. 108 (8), 1997-2006 (2015).
- Rastogi, K., Puliyakodan, M. S., Pandey, V., Nath, S., Elangovan, R. Maximum limit to the number of myosin II motors participating in processive sliding of actin. Scientific Reports. 6, 1-11 (2016).
- Debold, E. P., Patlak, J. B., Warshaw, D. M. Slip sliding away: Load-dependence of velocity generated by skeletal muscle myosin molecules in the laser trap. Biophysical Journal. 89 (5), 34-36 (2005).
- Albert, P. J., Erdmann, T., Schwarz, U. S. Stochastic dynamics and mechanosensitivity of myosin II minifilaments. New Journal of Physics. 16, (2014).
- Erdmann, T., Schwarz, U. S. Stochastic force generation by small ensembles of myosin II motors. Physical Review Letters. 108 (18), 1-5 (2012).
- Guo, B., Guilford, W. H. The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motility and the Cytoskeleton. 59 (4), 264-272 (2004).
- Miller-Jaster, K. N., Petrie Aronin, C. E., Guilford, W. H. A quantitative comparison of blocking agents in the in vitro motility assay. Cellular and Molecular Bioengineering. 5 (1), 44-51 (2012).
- Mansoon, A., Balaz, M., Albet-Torres, N., Rosengren, K. J. In vitro assays of molecular motors -- impact of motor-surface interactions. Frontiers in Bioscience. 13, 5732-5754 (2008).
- Persson, M., et al. Heavy meromyosin molecules extending more than 50 nm above adsorbing electronegative surfaces. Langmuir. 26 (12), 9927-9936 (2010).
- Kron, S. J., Spudich, J. A. Fluorescent actin filaments move on myosin fixed to a glass surface. Proceedings of the National Academy of Sciences of the United States of America. 83 (17), 6272-6276 (1986).
- Yanagida, T., Nakase, M., Nishiyama, K., Oosawa, F. Direct observation of motion of single F-actin filaments in the presence of myosin. Nature. 307 (5946), 58-60 (1984).
- Tsuda, Y., Yasutake, H., Ishijima, A., Yanagida, T. Torsional rigidity of single actin filaments and actin-actin bond breaking force under torsion measured directly by in vitro micromanipulation. Proceedings of the National Academy of Sciences of the United States of America. 93 (23), 12937-12942 (1996).
- Stewart, T. J., et al. Actin sliding velocities are influenced by the driving forces of actin-myosin binding. Cellular and Molecular Bioengineering. 6 (1), 26-37 (2013).
- Harada, Y., Sakurada, K., Aoki, T., Thomas, D. D., Yanagida, T. Mechanochemical coupling in actomyosin energy transduction by in vitro movement assay. Journal of Molecular Biology. 216 (1), 49-68 (1990).
- Fordyce, P. M., Valentine, M. T., Block, S. M. Advances in surface-based assays for single molecules. Single-Molecule Techniques: A Laboratory Manual. , 431-460 (2008).
- Ozeki, T., et al. Surface-bound casein modulates the adsorption and activity of kinesin on SiO2 surfaces. Biophysical Journal. 96 (8), 3305-3318 (2009).
- Neuman, K. C., Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nature Methods. 5 (6), 491-505 (2008).
- Neuman, K. C., Block, S. M.
- Thoresen, T., Lenz, M., Gardel, M. L. Thick filament length and isoform composition determine self-organized contractile units in actomyosin bundles. Biophysical Journal. 104 (3), 655-665 (2013).
- Matusovsky, O. S., et al. Millisecond conformational dynamics of skeletal Myosin II power stroke studied by high-speed atomic force microscopy. ACS Nano. 15 (2), 2229-2239 (2021).
- Reinemann, D. N., et al. Collective force regulation in anti-parallel microtubule gliding by dimeric Kif15 kinesin motors. Current Biology. 27 (18), 2810-2820 (2017).
- Cordova, J. C., et al. Bioconjugated core-shell microparticles for high-force optical trapping. Particle and Particle Systems Characterization. 35 (3), 1-8 (2018).
- Reinemann, D. N., Norris, S. R., Ohi, R., Lang, M. J. Processive Kinesin-14 HSET exhibits directional flexibility depending on motor traffic. Current Biology. 28 (14), 2356-2362 (2018).
- Forth, S., Hsia, K. C., Shimamoto, Y., Kapoor, T. M. Asymmetric friction of nonmotor MAPs can lead to their directional motion in active microtubule networks. Cell. 157 (2), 420-432 (2014).
- Shimamoto, Y., Kapoor, T. M. Analyzing the micromechanics of the cell division apparatus. Methods in Cell Biology. 145, 173-190 (2018).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Thoresen, T., Lenz, M., Gardel, M. L.
- Murrell, M., Thoresen, T., Gardel, M.
- Weirich, K. L., Stam, S., Munro, E., Gardel, M. L. Actin bundle architecture and mechanics regulate myosin II force generation. Biophysical Journal. 120 (10), 1957-1970 (2021).
