



Overview
ソース: ジョナサン・F・ブレイズ1,エリザベス・スーター1,クリストファー・P・コルボ1
1ワーグナーカレッジ生物科学科、1キャンパスロード、スタテンアイランドニューヨーク、10301
原核生物の定量的評価は、その豊富さ、指数関数的増殖の傾向、集団内の種の多様性、および特定の生理学的ニーズを考えると、厄介なことができます。この課題を複雑にすることは、細菌が複製する4相の性質(ラグ、ログ、静止および死)です。微生物の濃度を正確に推定する能力は、同定、単離、栽培、および特性評価を成功させるために必要である(6)。そのため、微生物学者は、臨床、産業、製薬、学術実験室環境(2,4,6)において、細菌およびウイルス負荷を確実に定量化するために、1世紀以上にわたってシリアル希釈および様々なめっき技術を採用してきました。この方法論の説明は、ドイツの科学者で医師のロバート・コッホが感染症を引き起こす薬剤に関する研究を発表した1883年に最初に現れました(2)。現代の細菌学の父と呼ばれることが多いコッホの前述の技術は、世界中の微生物の列挙のゴールドスタンダードとなっています。
シリアル希釈は、初期溶液(溶液0)を液体希釈剤(ブランク)の固定体積に連続的に再懸濁して、既知または未知の実体(溶質、生物など)の系統的な減少である。これらのブランクは通常0.45%の生理生理で構成されるが、組成物は変化させることができる(7)。実験者は、各希釈剤に対して任意の体積を選択できますが、ほとんどの場合、10の倍数であり、サンプルの対数減少を容易にします。例えば、溶液0は、10mLの栄養スープに懸濁した合計100個の大腸菌細胞を含む。溶液0の1mLを除去し、9 mLの生理生理物(希釈剤1)に添加した場合、新しい溶液(溶液1)は大腸菌の初期濃度の1/10分の1を含むであろう。この例では、新しい溶液(解1)には10の大腸菌細胞が含まれる。このプロセスを1mLの溶液1を除去し、別の9mLの生理塩分(希釈剤2)に添加することで、1つの大腸菌細胞のみを含む溶液2が得られます。各新しい溶液(希釈液9mL+溶液1mL)は合計10mLを含むので、この還元の希釈係数は10個であるか、またはこれが10倍の連続希釈であったと結論付けることができます(図1)。この例では 100 個のセルから始まり、10 倍の希釈を行っているため、1 セルの絶対最小濃度に達するには 2 つのステップのみが必要です。
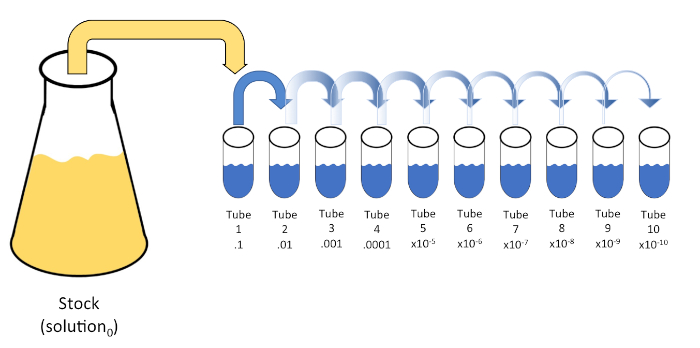
図 1: ストック ソリューションのシリアル希釈。ストック溶液(溶液0)の1 mLアリコートを、0.45%生理塩水の9 mLを含むチューブ1に添加する(希釈1)。この混合物の積は溶液1である。新しく作成した溶液1の1 mLをアリコートし、チューブ2に加えて繰り返します。アリクォートとリサスペンションは、最終的なチューブに達するまでこの方法で継続し、各ステップでそれぞれ10倍の10倍のストック濃度を希釈します。この図のより大きなバージョンを表示するには、ここをクリックしてください。
シリアル希釈は、所望の生物の管理可能な濃度を得るための最も簡単な技術であり、それは、微生物学者によって使用される多くのめっき技術のちょうど2つ、ペトリ皿のストリークと広がりによって補完されます。このアプローチのこの利点は、実験者が単一種の純粋な株を収穫したり、混合集団から別株を分離する(7)。ストリーキングは、適切な栄養素が利用可能な場合に成長する固体培地(一般的にアガロースからなる)に生物を導入することによって達成されます。硬い正弦波パターンで培地を通して無菌接種ループを静かに掃引し、実験者の波形の周波数に比例して生物を分配する。ペトリ皿を3分の1または4分の1(象限ストリーク)に分割し、皿の新しい領域が入力されるにつれて各ストリークの頻度を減らすことは、徐々にその領域を占有できる微生物の数を減らし、単一のコロニーを生成します。定量化できない細菌の芝生。スプレッドメッキは、サンプルを追加的に希釈しません。滅菌ガラス拡散機は、ペトリ皿全体に懸濁液媒体のアリコートを配布するために使用されます(図2)。スプレッドプレート上で成長するコロニーは、単一のセルから生じ、皿上の各コロニーは、CFU/mL(6)(図3)ソフト寒天およびレプリカとして表される所定の懸濁液中の1ミリリットル当たりのコロニー形成単位数を推定するためにカウントすることができる。めっきは、前述の技術のバリエーションであり、バクテリオファージおよび変異スクリーニングの分離を可能にする(1,7)。
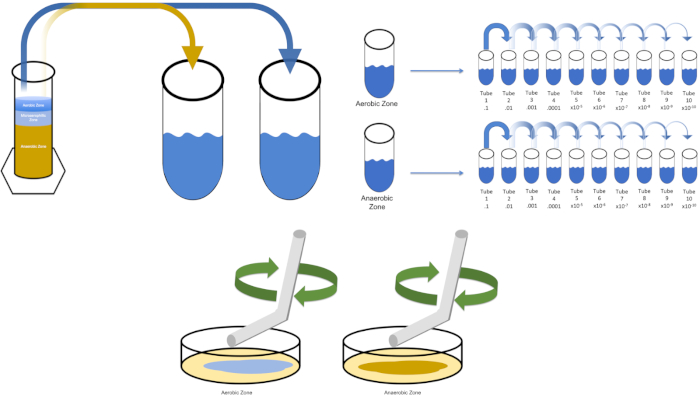
図2:細菌列挙および歪み分離のためのプレートストリーキング。ペトリ皿の底に識別情報(細菌名、日付、メディア)をラベル付けし、3分の1に分けます。ストックサンプルの適切な希釈を選択した後、無菌(使い捨てまたは炎)接種ループを取り、それを試験管(ここでは、T3)に沈めます。片側のペトリ皿カバーを少し上げて、接種ループだけが寒天にアクセスできるようにします。寒天を危険にさらさないために注意しているジグザグの方法でメディアの上部を横切って接種ループを滑空します。プレートを約1/3rd(〜118°)回転させ、ジグザグ運動の周波数を下げます。最後の時間を回転させ、ジグザグの周波数をもう一度下します。この図のより大きなバージョンを表示するには、ここをクリックしてください。
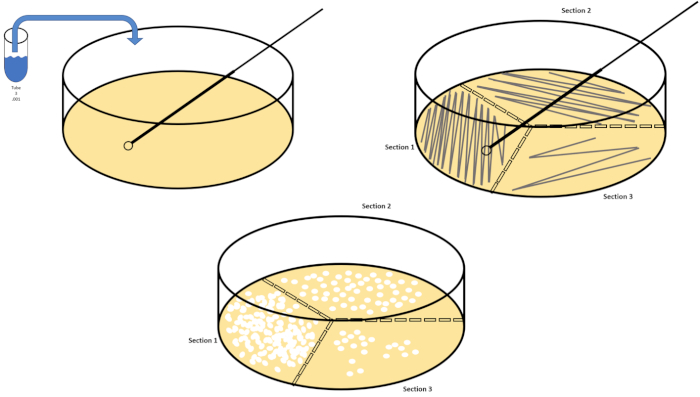
図3:スプレッドメッキ。好気性ゾーンの1gをT1で再懸濁し、次いで連続的に希釈した。滅菌ガラスまたはプラスチックの使い捨て可能な広がる棒は各皿に接種を配るために使用される。これを嫌気性ゾーンの1gで繰り返した。この図のより大きなバージョンを表示するには、ここをクリックしてください。
連続希釈と同様に、対数スケールは、組織の濃度を表すために使用されます。100mm x15mmを測定する標準的なペトリ皿で成長したコロニーの数は、成長の孤立したクラスターを識別することによって手動で(または計算処理の助けを借りて自動化される)列挙することができる。合計が 30 未満または 300 未満のカウントは、それぞれカウントする数が少なすぎる (TFTC) またはカウントが多すぎる (TNTC) と定義する必要があります。後者の場合、新しいペトリ皿を再ストリークする前に濃度を下げるシリアル希釈を行う必要があります。3つの別々のペトリ皿から識別された自己完結型コロニーの数を平均し、希釈係数によって平均を乗算すると、CFU/mLが得られます。CFU/mLのログ10を時間に対してプロットすると、生物の平均生成時間が明らかになる(7)。
Procedure
1. セットアップ
- すべての材料、段階的な実験プロトコル、および消耗品を廃棄する方法を記載したフローチャートは、実験室のノートブックに書かれ、実験用ワークスペースの近くに保管する必要があります。
- ワークスペースは、適切な消毒剤(70%エタノール)で殺菌する必要があり、実験者はまた、暴露異常からそれらを保護するきれいな実験室の衣服を着用することにより、汚染リスクを軽減する必要があります。適切な衣服には、ラボコート、ラテックスまたはニトリル手袋、グーグル、人工呼吸器、クローズドシューズが含まれますが、これらに限定されません。常に無菌技術を維持することが重要です。
- 0.45%生理生理の90 mLを調出します。きれいな段階的なシリンダーを使用して、無菌水の90 mLを測定し、0.45%の生理生理知とラベル付けされたきれいなアーレンマイヤーフラスコに移す。塩化ナトリウムの.405 g(シグマアルドリッヒNaCl S9888)の重量を量り、0.45%の生理食塩水を標識したフラスコに加えます。溶質が見えなくなるまで繰り返し旋回します。
- 完了すると、実験者はすべての表面を再殺菌し、OSHAガイドラインに従って不要な生物、希釈株、ペトリ皿、または使い捨て接種ループを廃棄する必要があります。実験室の衣類は手を洗う前に取除くことができる。
2. メディアの準備
- 所望の生物の栽培に適した培養媒体を選択します。ほとんどのシナリオでは、スープは十分な細菌の増殖を可能にします。ウィノグラツキープロトコルからの生物がここで望まれているので、炭酸カルシウム、硫黄、セルロース、泥からなるカラムを組み立て、7日間邪魔されずに残しました。前命名のカラムは、好気性、好好気性、嫌気性のセクションに分離されています。
- 対象の生物をめっきするのにふさわしい媒体を選ぶ。微生物学グレードの寒天を用いる液体媒体の補充は、典型的には固化剤として用いられる。LB培地/寒天は、前名称のカラムの好気性、好好気性および嫌気性領域からのサンプルを収穫する場合に十分である。注:この手順では、マイクロエアロフィルリック領域からのサンプルは採取されなかった。しかし、これらの生物は、ろうそく瓶で栽培する必要があります。シール前にこの栽培室にろうそくを導入すると、マイクロ好気性増殖に適した低酸素環境を作成します。
- 250 mLを用意したいので、オートクレーブ時のボイルオーバーを防ぐために500mL(またはそれ以上)のアーレンマイヤーフラスコを使用してください。1つは「ブロス」、もう1つは「寒天」とラベルを付けます。
- 製造元の濃度に関する推奨事項に従って、各ソリューションの作成に必要なメディアの量を決定します。ここで使用されるLB寒天は、25g/Lと超純水を組み合わせて調製します。250 mLの私たちの容積は6.25 LB寒天/250 mLの水の解決を要求する。同様に、LBブロスと水の同じ比率を組み合わせて調製されます。固化剤で補わないため、冷却しても硬化しません。
- メディアの重量を量り、メーカーの推奨事項と一致する割合で水と混合します。「寒天」と書かれたフラスコに6.25gのLB寒天を加え、LBブロスの6.25gを「ブロス」と書かれたフラスコに加えます。各フラスコに250mLの超純水を加えます。
- 各フラスコにアルミホイルをラップし、オートクレーブを使用して、121°C、15 psiで最低15分間メディアを殺菌します。
- 耐熱手袋またはパッドを使用して、サイクルが完了したらオートクレーブからフラスコを取り外し、40-50°Cの水浴に入れます。
- 適切な温度に達したら、250 mLアーレンマイヤー、または丸底フラスコに「ブロス」とラベル付けされたフラスコの内容物を注ぎます。250 mLフラスコ「溶液0」にラベルを付けます。
- 10、100mm x 15 mmの滅菌ペトリ皿を取得し、日付、名前、使用されるメディアの種類、および生物が収穫されるウィノグラツキーカラムゾーンでラベルを付けます。
- 水風呂から「寒天」と書かれたフラスコを取り出し、10個のペトリ皿に注ぎ始めます。各料理に15mL以下を追加する必要があります。これはまた、精度を向上させるためにピペットおよび25 mLの血清ピペットで行われてもよい。滅菌ピペットチップを使用して、存在する気泡を除去し、プレートの蓋で覆い、一晩固化するために残します。
3. 希釈剤の調製
- ラックに20mL以上を収納できる試験管を10本用意し、T1-T10にラベルを付けます。各チューブ番号は、それが対応する希釈係数と一致しています(すなわち、T4 = 1x10-4または0.0001またはストック濃度の1/10,000分の1)。
- 10の試験管のそれぞれに0.45%の生理物のピペ9 mL。
- 生理食生のブランクは今オートクレーブによって殺菌される準備ができている。アルミホイルを使用して10個の試験管をそれぞれカバーし、オートクレーブ互換試験管ラックに移します。121°C、15 psiで最低15分間殺菌します。
- 耐熱手袋を使用してブランクを取り除き、冷却することができます。チューブが室温に達したとき、またはタッチに冷えるとき、必要になるまで4°Cでカバーして保管してください。
4. 対象生物の育成
- 以前に縞が付いたプレートまたは凍結ストックの50 μLから単一のコロニーで「溶液0」を接種する。接種された「溶液0」を一晩振って37°Cのインキュベーターに入れて、標的生物に複製する時間を与えます(必要に応じて)。(注:フラスコは汚染を防ぐために覆われるべきです。対象生物が好気性の場合は、無菌ガーゼと綿栓を使用して汚染を防ぎます。ウィノグラツキーカラムの領域を評価する場合は、単に各所望のゾーンから1グラムを削除し(この研究の目的のために好気性と嫌気性)、ステップ5.3に進む前にT1で再中断します。
5. シリアル希釈
- インキュベーターから「栄養スープ」と表示されたフラスコを入手し、激しく振ります。
- 「溶液0」のピペ状1mLをT1標識の試験管に入れた。渦 T1.ウィノグラツキーの移植を評価する場合は、所望のゾーンの1グラムの重量を量り、渦の前にT1に追加します。(注:1 mLは、単純化のために使用され、より小さい量の希釈剤も使用してもよい)。
- 試験管T1から1 mLを取り出し、試験管T2に追加します。渦 T2.
- 試験管T2から1 mLを取り出し、試験管T3に追加します。渦 T3.
- 試験管T3から1 mLを取り出し、試験管T4に追加します。渦 T4.
- 試験管T4から1 mLを取り出し、試験管T5に追加します。渦 T5.
- 試験管T5から1 mLを取り出し、試験管T6に追加します。渦 T6.
- 試験管T6から1 mLを取り出し、試験管T7に追加します。渦 T7.
- 試験管T7から1 mLを取り出し、試験管T8に追加します。渦 T8.
- 試験管T8から1 mLを取り出し、試験管T9に追加します。渦 T9.
- 試験管T9から1 mLを取り出し、試験管T10に追加します。
6. スプレッドメッキ
- T1からペトリ皿に直接希釈されたサンプルのピペ100 μL。このステップは、チューブごとに繰り返すことができますが、必要はありません。
- 無菌の使い捨て拡散棒を得るか、または炎はガラス拡散棒を殺菌する。時計回り/反時計回りの動きで、広がるロッドの水平部分を滑空して、ペトリ皿内にサンプルを均等に分配します。
- 評価する Winogradsky 列のゾーンごとに繰り返します。
- 37°Cのインキュベーターでプレートを24時間インキュベートします。嫌気性生物の場合は、嫌気性チャンバーを使用してください。
7. ストリーキング
- ターゲット生物の適切な希釈を選択します。例えば、溶液4は、初期濃度の1/10,000分の1の希釈をもたらす。典型的には、1/1,000th(T3/solution)、1/1,000,000th(T6/Solution6)および1/1,000,000,000番目(T9/Solution 9)の希釈が評価され、微生物を列挙する。
- プラスチック無菌使い捨て接種ループまたは10秒以上の間火の下にあった再利用可能な金属接種ループを使用して、ステップ5から所望の溶液に浸漬する。キャリブレーションされた接種ループは0.01 mLを転送する必要があります。(注意:熱から取り除く直後に炎ループが細菌に接触しないようにする)
- 片側のペトリ皿カバーを少し上げて、接種ループだけが寒天にアクセスできるようにします。寒天を危険にさらさないために注意しているジグザグの方法でメディアの上部を横切って接種ループを滑空します。ペトリ皿のふたを下ろします。
- 新しい使い捨て可能な接種ループを使用するか、再利用可能なループを再殺菌します。
- プレートを約1/3rd(〜118°)回転させ、ジグザグ運動の周波数を下げます。
- 繰り返しますが、新しい使い捨てループを使用するか、最終時間を回転させる前に金属ループを再殺菌し、ジグザグ周波数をもう一度減らします。ペトリ皿のふたを下ろします。
- 新しい使い捨てループを使用するか、再利用可能なループを再燃やすことによって、少なくとも3つのペトリ皿が3つの異なる希釈のために縞模様になるまで、手順7.2-7.6を繰り返します(図2)。
- ストリークペトリ皿を一晩37°Cのインキュベーターに入れます。嫌気性生物の場合は、無気性チャンバーを使用します。
8. データ分析と結果
- 培養物は、7日間のウィノグラツキー列の酸化ゾーンと無酸化ゾーンから採取した。これらのゾーンは、それぞれヘテロ栄養および鉄酸化性アナエロベに適しています。
- カラムの移植は、LB寒天プレート上で縞または広がる前に連続的に希釈された。
- ストリーキングは、評価されたウィノグラツキーゾーンのそれぞれから混合集団を明らかにしました。スプレッドプレートは同様の結果を生み出した。
- CFU/mL または CFU/g を計算するには、3 つのプレートからカウントされたコロニーの数を平均します。コロニーの平均数に希釈係数を掛け、引用符で囲まれた量で除算します。例えば、溶液6(T6)の0.1 mLを接種したプレート上で平均65のコロニーをカウントした場合、前述の式は650,000,000 CFU/mLに等しくなります。
- 分離されたコロニーは、種のアイデンティティを決定するために濃縮アッセイで使用するために、各プレートから選択できるようになりました。
時には、細菌を同定して研究するためには、まずサンプルからそれらを分離し、濃縮する必要があります。例えば、ウィノグラツキーカラムから得られたサンプルは混合され、複数の種または細菌の株が含まれているため、個々の細菌を研究したり、存在する異なる種類を列挙することは困難な場合があります。この目的のために、シリアル希釈およびめっき技術は、通常、細菌負荷を確実に定量し、個々のコロニーを分離するために使用される。
シリアル希釈は、この例における細菌である生物の濃度が、液体希釈液の固定量における連続的な再懸濁を通じて系統的に減少するプロセスである。通常、希釈体の体積は、サンプル生物の対数減少を容易にする10の倍数である。例えば、1グラムの堆積物は、まず目的のウィノグラツキーゾーンから除去され、適切な液体媒体の10ミリリットルに添加される。次に、この第1希釈の1ミリリットルを、培地の9ミリリットルを含む別のチューブに添加する。このプロセスは、いくつかの異なる濃度の細菌が調製されるまで繰り返すことができる。この例では、ウィノグラツキーカラムからの混合サンプルには未知の、しばしば大きな数の細菌が含まれているため、シリアル希釈は細菌の列挙の鍵となります。
次に、ストリークメッキとスプレッドメッキにより、サンプル内の細菌の単離と列挙が可能になります。ストリーキングは、3分の1に分けられる栄養素を補充した固形培地の1つのセクションに希釈されたサンプルを導入することによって達成される。この接種は、ジグザグパターンでプレートの各3分の1に広がります。プレートの異なるセクションが縞模様化され、前のサンプルから一度だけ交差すると、サンプルはより薄く広がります。つまり、後のセクションで個々のコロニーを達成するために、1つの希釈からストリークするだけで済む場合があります。インキュベーション後、縞模様のプレートは、コロニー形態の観察を可能にし、異なる細菌種を区別するのに役立つ情報を提供する。
あるいは、主な目的が試料拡散めっきにおける細菌の列挙である場合に使用してもよい。スプレッドメッキでは、単一のサンプルのアリコートが固体媒体の表面全体に均等に広がります。通常、混合サンプル中の細菌数がわからないため、各希釈またはそれらの代表的なサンプルに対してスプレッドプレートが作られます。インキュベーション後、これらのスプレッドプレートを用いて列挙を行うことができる。コロニー数が 30 未満のプレートは、小さいカウントが大きな誤差の影響を受けるため、破棄する必要があります。同様に、コロニーの混雑と重複がコロニー数の過小評価につながる可能性があるため、300を超えるカウントは破棄する必要があります。これらの残りの各料理のコロニー数を記録し、希釈係数を掛けてから、体積めっきで割ると、コロニー形成単位(CFC)が懸濁液1ミリリットル当たり生成されます。このビデオでは、既知の細菌を含むサンプルと、シリアル希釈、スプレッドメッキ、および筋めっきを使用してウィノグラツキーカラムの様々な領域に含まれる微生物群を定性的かつ定量的に評価する方法を学びます。
まず、ラボコート、手袋、ゴーグルなどの適切な個人用保護具を着用してください。次に、70%のエタノールでワークスペースを殺菌し、表面を拭き取ります。次に、2つの500ミリリットルのアーレンマイヤーフラスコを収集し、1つのスープと他の寒天にラベルを付けます。LB寒天溶液を調製するには、約6.25グラムのLB寒天、3グラムのテクニカル寒天、および寒天とラベル付けされたフラスコに蒸留水の250ミリリットルを混ぜます。
次いで、2を組み合わせてLBスープを調作する。5グラムのLB培養物と100ミリリットルの蒸留水をフラスコにラベル付けしたスープに入れます。フラスコをオートクレーブした後、耐熱手袋を使用してオートクレーブからフラスコを取り除き、40~50°Cの水浴に入れます。フラスコが摂氏50度になったら、スープ溶液の100ミリリットルのアリコートを3つ慎重に準備し、各アリコート溶液にゼロのラベルを付けます。次に、10の滅菌ペトリ皿を収集し、生物が収穫される日付、名前、使用されるメディアの種類、およびウィノグラツキーカラムゾーンでそれらをラベル付けします。寒天フラスコから各ペトリ皿に寒天のピペット15ミリリットル。その後、ピペットの先端を使用して気泡を除去し、プレートの蓋を交換し、一晩ベンチトップに固められるようにします。
翌日、70%のエタノールでベンチトップを拭きます。次に、T1からT10までの10 20ミリリットルの試験管にラベルを付け、ラックに入れます。各チューブに.45%生理食塩水のピペット9ミリリットル。次に、10個の試験管をそれぞれキャップでゆるく覆い、オートクレーブ対応の試験管ラックに移します。サイクルが完了したら、耐熱手袋を使用して生理食生のブランクを取り除き、冷却することができます。管は約22°Cに達するまで室温で保管してください。
既知の標的生物を培養するために、この例では大腸菌を、以前に縞模様のプレートから単一のコロニーで溶液ゼロの100ミリリットルを接種する。その後、チューブをカバーし、摂氏37度で一晩それをインキュベートします。ウィノグラツキーカラムの領域を評価するには、好気性ゾーンからT1に約1グラムの材料を追加し、渦によって再中断します。次に、嫌気性ゾーンから1グラムの材料でこのプロセスを繰り返します。
大腸菌を接種した溶液を含むチューブをインキュベーターから取り出し、振ります。次に、溶液の1ミリリットルをT1試験管と渦に混合して混合する。T1から溶液の1ミリリットルを取り出し、それをT2に移し、混ぜ合わせます。チューブT10を通してこのプロセスを繰り返します。ウィノグラツキーカラムの好気性および嫌気性ゾーンを評価するには、以前に調製したT1チューブから1ミリリットルの溶液を取り出し、適切なT2チューブに移します。次に、先に示したようにT10管を通してシリアル希釈を続ける。
プレートを広げるには、各T3チューブから対応するペトリ皿に希釈されたサンプルのピペット100マイクロリットル。次に、無菌の広がり棒を使用して、サンプルをペトリ皿に穏やかに分配し、プレート蓋を交換します。前に示したように、T6 および T9 希釈に対してこのプロセスを繰り返します。好気性生物を含むプレートを37°Cのインキュベーターで24時間インキュベートします。嫌気性の室内で嫌気性生物を含むプレートを24時間摂氏37度に設定します。翌日、インキュベーターと嫌気室からT3、T6、T9希釈プレートを取り出し、ベンチトップに移します。一度に1つのプレートで作業し、ジグザグパターンでメディアの上部を横切って無菌接種ループを滑走します。次に、ペトリ皿の蓋を交換します。次に、プレートを1/3回転させ、ループを殺菌して、以前に作られたジグザグパターンの周波数を減らします。繰り返しになりますが、ループを殺菌した後、プレートを1/3回転させ、ジグザグパターンの周波数を最後に1回減らし、蓋を交換します。前に示したように、残りのプレートに対してこのストリーキング方法を繰り返します。次に、好気性生物を含む縞板を一晩37°Cのインキュベーターに入れ、嫌気性の生物を含む縞板を一晩37°Cに設定します。
文化は7日間のウィノグラツキーコラムの好気性および嫌気性ゾーンから収穫された。その後、培養物を連続的に希釈し、LB寒天プレートに広げた。ストリーキングは、評価されたウィノグラツキーゾーンのそれぞれから混合集団を明らかにし、スプレッドプレートは同様の結果を生み出しました。混合集団から縞模様のプレートは、異なる形状、サイズ、テクスチャ、および色の細菌コロニーになります。対照的に、既知の生物大腸菌を含む縞および広がりプレートは、相同集団を示した。一般に、同じサンプルと希釈係数で広がる3つのプレートの平均コロニー数を使用して、1ミリリットル当たりのCFCを計算することをお算するのが最善です。コロニーの平均数に希釈係数を掛け、引用符で囲まれた量で除算します。最後に、各プレートから選択された単離されたコロニーは、種のアイデンティティを決定するために、さらなる濃縮アッセイに使用することができる。
Applications and Summary
メッキによる細菌の列挙およびひずみ分離には、標的生物の管理可能な濃度が必要です。従って、成功しためっきは連続希釈に起因する。したがって、前述の技術は、微生物学的検査および実験の基礎であり続ける。設計上簡単ですが、希釈因子とめっき技術は、各方法の完全性を損なうことなく結果を強化するために実験者によって変更することができます。細菌の増殖の4つの段階をプロットすることは、所望の微生物を特徴付けるときに役立ちます。これらの相、ラグ、ログ、静止、および死は、細菌複製の変化によって特徴付けられます。遅れ期は、生理的適応による成長が遅く、ログ相は生存細胞の指数関数的上昇を特徴とする最大増殖の期間であり、その後、環境の制限と毒素の蓄積のために静止期に達し、細胞数が減少し始める死期の前に。これは、連続的に希釈(または混乱を避けるために1ステップの希釈)によって達成することができ、時間0から始まる合計8時間の1時間ごとに解対溶0(解対0は、各希釈後に振盪インキュベーターに戻す必要があります)。時間0の単一希釈体の CFU/ml のログ10を計算し、Y 軸にプロットします。サンプル時間1に対してこの計算を繰り返します (時間0と同じ希釈係数を使用して CFU/mL を計算してください)。X 軸に毎回(時間1-時間8)がプロットされるまで繰り返します。
References
- Allen, M.E., Gyure, R.A. (2013) An Undergraduate Laboratory Activity Demonstrating Bacteriophage Specificity. Journal of Microbial Biological Education 14: 84-92.
- Ben-David, A., Davidson, C.E. (2014) Estimation Method for Serial Dilution Experiments. Journal of Microbiological Methods 107:214-221.
- Goldman, E., Green, L.H. (2008) Practical Handbook of Microbiology.
- Koch, R. (1883) New Research Methods for Detection of Microcosms in Soil, Air and Water.
- Lederberg, J., Lederberg, E.M. (1952) Replica Plating and Indirect Selection of Bacterial Mutants. Journal of Bacteriology 63:399-406
- Pepper, I., Gerba, C., Ikner, L. (2019) Bacterial Growth Curve Analysis and its Environmental Changes. JoVE Science Education Database. Environmental Microbiology.
- Sanders., E.R. (2012) Aseptic Laboratory Technique: Plating Methods. JoVE 63:e3063.