计算机准则的基础结构和功能的蛋白质使用的I - TASSER管道的特性描述。开始,从查询蛋白质序列生成三维模型使用多个线程的路线和迭代结构装配模拟。此后绘制功能的推论是基于匹配与已知的结构和功能的蛋白质。
Method Article
基于计算机的蛋白质结构与功能预测议定书
Opens in a new tab
In This Article
Summary
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Abstract
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
基因组测序项目,以加密形式以百万计的蛋白质序列,这就要求它们的结构和功能的知识,提高其生物作用的认识。虽然实验方法提供了这些蛋白质的一小部分的详细信息,计算模型所需要的大多数实验未知的蛋白质分子。我TASSER服务器是一个高分辨率的蛋白质结构与功能的建模上线的工作台。由于蛋白质序列,从我的TASSER服务器上的一个典型输出包括二级结构预测,预测每个残基的溶剂可及,同源模板线程和结构对齐检测蛋白质,最多五个全长高等教育的结构模型,和基于结构酶的分类,功能注释基因本体术语和蛋白质 - 配体结合位点。所有的预测标记的信心分数讲述如何准确的预测是不知道的实验数据。为了方便最终用户的特殊要求,的服务器提供渠道接受用户指定的残留间的距离和联系映射到交互地改变我TASSER建模,而且还允许用户指定的任何蛋白质为模板,或排除任何模板蛋白质在结构装配模拟。结构信息可以收集用户基于实验证据或生物的见解与我TASSER预测的质量改善的目的。服务器被评为蛋白质的结构和功能预测,在最近社会广泛CASP实验的最佳方案。目前有20000注册来自100多个国家的科学家使用的是上线的我,TASSER服务器。
Protocol
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
方法概述
序列结构功能的范例之后,我TASSER程序的结构和功能建模 1-4涉及四个连续的步骤:(一)LOMETS 5模板识别;(二)重组由副本的片段结构交流蒙特卡罗模拟6;(C)原子水平结构使用REMO 7和8 FG - MD的细化;及(d)基于结构的功能解释使用的辅助因子9。
模板识别 :对于用户提交的查询序列,该序列是先穿过一个代表性的PDB结构库由本地安装LOMETS元线程服务器。线程是用于识别模板的蛋白质可能有类似的结构,或包含类似的查询蛋白质的结构图案的序列结构调整过程。为了增加覆盖同源templ吃了检测,LOMETS结合多个国家的最先进的算法,涵盖不同的线程方法。由于不同的线程方案有不同的评分系统和校准灵敏度,质量评估从每个线程程序生成的线程的路线归Z - score模型 ,其定义为: 
Z - score模型是由程序生成的所有路线的统计平均得分相对标准差单位;和Z 0,是一个具体方案Z - score模型的截止确定大规模的线程基准测试的基础上,区分"好"和"坏"的模板。高的Z - score模型的模板意味着顶部的模板对齐得分明显比大多数其他的模板,这通常意味着,对齐对应一个很好的模式。如果顶部的线程模板最有嗨GH归Z分数 ,最终我TASSER模型的准确性通常很高。但是,如果蛋白质是大型线程路线的覆盖范围是局限于一个小区域查询蛋白,高标准化的Z -得分并不一定意味着全长模型的建模精度高。前两名从每个线程程序的线程路线是收集和使用,为下一步的结构组装。
迭代结构装配仿真 :线程程序,查询的顺序是线程对齐和未对齐的地区分割。从模板线程对齐的连续片段切除,并直接用于结构装配,而从头建模建未对齐的循环区域。副本交换蒙特卡罗模拟6引导晶格系统结构大会议事执行。我的TASSER力场包括氢波nding相互作用10,以知识为基础的统计能量从已知的蛋白质结构的 PDB 11 日 ,SVMSEQ 12的序列为基础的接触预测 ,并从5 LOMETS线程模板收集空间限制的条款。由SPICKER 13日聚集在低温副本过程中产生的模拟的构象诱饵确定低自由能态的结构。顶部集群的集群重心得到平均的所有群集结构诱饵的三维坐标和用于最终模型生成。消除立体冲突,并进一步完善了全局拓扑,模拟和聚类过程重复两次。
原子级模型的建设和完善 :群集SPICKER聚类后得到的质心降低蛋白质模型(代表其 Cα和侧链的重心每个残基)和HAVE有限的生物应用。从减少模型的建设是全原子模型分两步进行。第一步,REMO 7用于全原子模型,通过优化的氢键网络构建的C -α的痕迹。在第二个步骤,REMO全原子模型进一步细化的FG - MD 14,提高骨干的扭转角,键长,侧链rotamer方向,通过分子动力学模拟,作为指导从搜查的结构片段商标由PDB结构调整。 FG - MD的成品模型被用作最后的三级结构预测模型,我的TASSER。
所生成的模型的质量估计基础上的信心得分(得分),其定义是基于LOMETS线程路线的Z得分和我TASSER模拟的收敛,数学制订: 
其中13日确定的结构性集群结构诱饵的多重性; 中号TOT是提交到集群的诱饵总数; 聚集诱饵集群重心平均的RMSD; Norm.Z分数(I)的归Z - score模型(公式1) 我在LOMETS 5个线程的服务器获得的顶级线程对齐; N是LOMETS使用的服务器数量。
这架C -得分有很强的相关性与我TASSER模型的质量。得分的C和蛋白质的长度相结合,首先,我TASSER模型的精确度可估计平均误差为0.08 TM得分和2的RMSD 15 Å。在一般情况下,车型的C -评分> - 1.5预计将有一个正确的折叠。在这里,RMSD值和TM -得分都是众所周知的模型和原生结构之间的拓扑相似的措施。 TM -得分VALUES在[0,1]范围内,更高的分数表明,更好的结构匹配16,17。然而,排名较低的车型(即2 日 -5日模型),与TM -评分和RMSD的C -得分的相关性要弱得多(〜0.5),不能用于模型的质量绝对可靠估计。
第一种模式,总是在我TASSER模拟的最好的模式?这个问题的答案取决于目标类型。容易攻击的目标,第一个模型是通常的最佳模式,其C -分数通常比其他模型高得多。然而,硬性指标,线程不会有显著的模板点击,第一种模式是不一定的最佳模式,我的TASSER实际上已经难以选择最好的模板和模型。因此,建议为硬性指标来分析所有的5款车型,并选择他们的实验信息和生物知识为基础。
功能PREDictions:在最后一步,从FG - MD的最终生成三维模型,用于预测蛋白质功能的三个方面,即:一)酶委员会(EC)18和(b)基因本体(GO)的19个条款和( C)为小分子配体结合位点。所有三个方面,产生功能的解释是使用辅助因子,这是一个新的方法来预测蛋白质的功能,基于全局和局部相似模板蛋白与已知的结构和功能的PDB的。首先,全球预测模型的拓扑匹配功能的模板库使用结构对齐程序TM -对齐20。下一步,蛋白质最相似的目标模式选择基于其全球结构的相似性的图书馆,进行识别附近的主动/结合位点区域的结构和序列相似性和广泛的本地搜索是。由此产生的全球和本地的相似分数排名模板蛋白(功能同系物)和转移的注释(EC编号和基因本体19条款)的基础上顶尖的得分命中。同样,配体结合位点残基与配体结合模式的推断基础上的局部比对查询已知的配体结合位点残基在顶部的打分函数模板9。
功能质量(教统会和好长期)我TASSER预测评估的基础上功能的同源性得分(FH -得分),这是一个全球和地方之间的查询和模板的相似性的措施,并定义为: 
其中C -评分是估计式中所界定的预测模型的质量。 (2); TM得分措施之间的全球模型和模板蛋白结构相似性的RMSD 阿里是在TM -对齐20的结构对齐地区之间的模型和模板结构的RMSD;病毒代表结构对齐(即除以查询长度的结构对齐残留的比例)的覆盖面 ; ID阿里的TM -对齐对齐序列标识。估计EC数量预测的信心得分也包括任期内定义的局部地区,计算评估查询和模板之间的活跃站点匹配(ACM): 
其中 ,N t表示模板残留在当地的数量,N阿里是对齐的查询模板残留对数 ,D II是我日对对齐残留的 Cα之间的距离 ,D 0 = 3.0距离截止,M II是第i个对对齐残留之间的BLOSUM分数。在一般情况下,FH -得分的范围是[0,5]和ACM的得分是在[0,2],其中分数越高,表明功能分配更加自信。 ACM得分也可用于评价局部结构和序列相似性配体结合位点,这被称为BS得分附近。
1。提交蛋白质序列
- 访问在我的TASSER网页http://zhanglab.ccmb.med.umich.edu/I-TASSER开始的结构和功能模拟实验。
- 复制并粘贴到所提供的氨基酸序列,或直接上传到您的计算机,通过点击"浏览"按钮。我TASSER服务器目前接受高达1500残基序列。超过1500残留的蛋白通常是多结构域蛋白,并建议将分裂成单独的域,然后提交到我的TASSER。
- 提供您的e - mail地址(强制性)和工作(可选)的名称。
- 用户可以选择指定外部RESidue接触/距离限制,添加一个额外的模板或排除在结构建模过程中的一些模板蛋白。了解更多有关使用这些选项中的"讨论"一节的。
- 要提交的序列中,单击"运行我的TASSER"按钮。该浏览器将被定向到一个确认页面,显示用户指定的信息,作业标识(作业ID)号码和一个链接到网页的结果将存入作业完成后。用户可能书签此链接或记下来,以供将来参考的作业标识号。
2。结果的可用性
- 检查您提交的作业状态http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php ,在我TASSER队列页访问。点击"搜索"选项卡和使用作业身份证号码或查询序列搜索您提交的作业。
- 莫后的结构和功能deling完成后,通知电子邮件包含图像的预测结构和网络链接将发送给您。点击此链接或打开在步骤1.5中的链接,书签,浏览和下载的结果。
3。二级结构和溶剂可预测
- 检查结果页的顶部显示FASTA格式的查询序列。如果任何额外的约束/模板指定的序列提交时,网页显示用户指定的信息的链接,也可以看出(图1A)。
- 检查显示为:(H)的α-螺旋,β链(S)或线圈(C)和预测每个残基的信心得分(0 =低,9 =高)的二级结构预测。寻找地区很长一段普通中学结构(H或S)预测,估计中的蛋白质的核心区域。二级结构元素的分布的基础上,还可以分析蛋白质结构类。铝因此,线圈元件的地区中的蛋白质通常表明非结构化/无序的地区。
- 查看预测溶剂,以确定埋和溶剂暴露在查询区域的无障碍(图1C)。值预测溶剂无障碍范围从0(埋残留)到9(暴露残留)。大多埋藏区包含残留,可用于划定核心区域中的蛋白质,而溶剂暴露和亲水性残留物的地区是潜在的水化/功能网站。
4。三级结构预测
- 向下滚动以查看预测查询蛋白质的三级结构,小程序(图2)在互动Jmol显示。在这个程序上左单击要更改显示结构的外观,放大到特定区域,选择特定的残留物类型,在预测模型或计算残留间的距离。
- 分析模型长非结构化地区的存在。这些řegions通常对应到蛋白质的无序地区或表明缺乏模板对齐。这些地区一般都具有建模精度低,在从N&C -末端区域将提高建模精度建模和消除这些地区。
- 点击"下载模式"链接,下载模型的PDB格式的结构文件。在任何分子可视化软件(如Pymol,Rasmol等)的结构特征的进一步分析,你可以打开这些文件。
- 估计预测结构的质量,分析结构模型的信心得分(C得分)。通常的C -评分值(公式2)范围内的[-5,2],其中一个更高的分数反映了质量更好的模型。估计TM -第一款车型的得分和RMSD值显示为"模型1的估计精度"。对于长期的蛋白质,它是建议基于TM得分的评估模型的质量,TM得分比的RMSD的拓扑变化更加敏感。 <李>点击链接分析C -得分,集群规模和集群密度的所有型号的"关于C -分数"。预计TM得分和RMSD是仅适用于第I - TASSER模型,因为的C -得分排名较低的车型不是强与TM -得分或RMSD相关。集群密度和集群规模相对第一种模式的基础上,可以部分评估排名较低的车型质量,其中,从更大的集群和高密度平均接近天然结构的模型。
- 低C -得分的预测通常表明低精度的预测。在大多数这种情况下,查询蛋白质缺乏良好的模板和库中的大小从头建模(即> 120残留)的范围之外。在这种情况下,用户可以寻找额外的空间限制,并用它们来改善我TASSER建模(见讨论部分)。它也鼓励我们的夸克服务器提交的序列(夸克/"> http://zhanglab.ccmb.med.umich.edu/QUARK/)为纯从头建模,如果蛋白质的大小低于200残留。
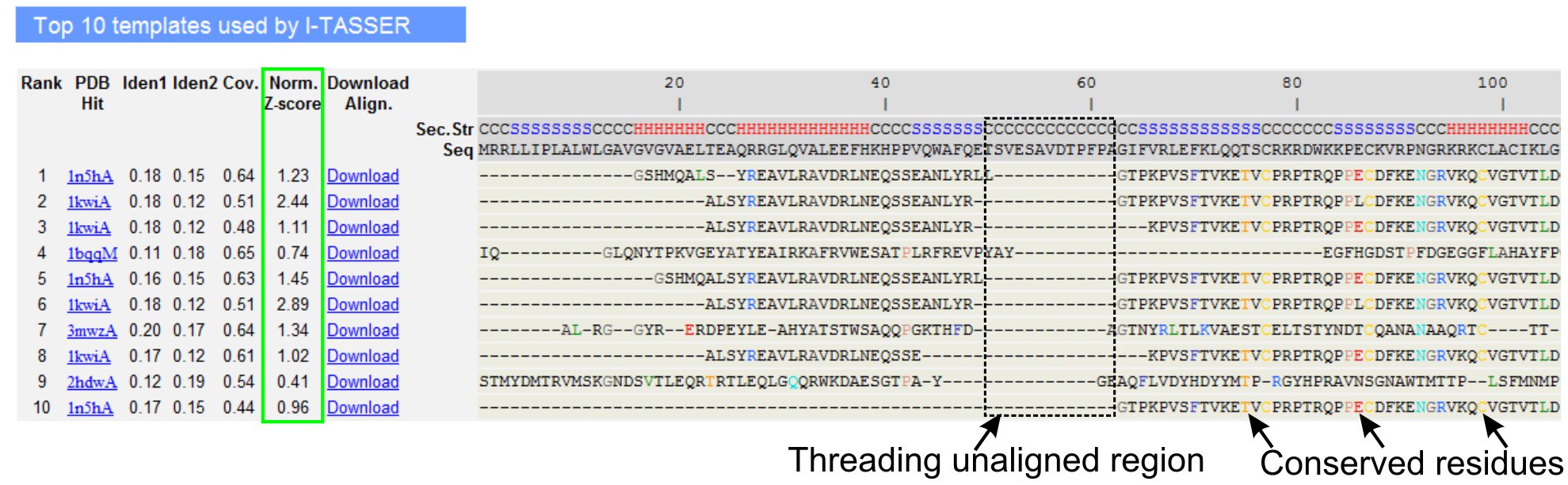
5。 LOMETS目标模板对齐
- 向下滚动分析十大线程模板查询的蛋白质,LOMETS线程程序(图3)确定。查看归一化的Z -得分(公式1),在"规范。 Z - score模型"一栏中,分析线程对齐质量。一个规范化的Z - score模型> 1的比对,体现了一种自信的路线和最有可能作为查询的蛋白质相同的折叠。
- 在线程对齐的地区(1列"IDEN。")和整个供应链(列"IDEN 2)评估之间的查询和模板蛋白的同源性分析序列的身份。高的同源性查询和模板蛋白质之间的进化关联的指标。
- 查看线程对齐彩色显示的残留物,视觉识别优劣erved残留/查询和模板蛋白的图案。一个线程对齐地区较高的同源性相比,全链对齐也表示保守的结构基序/查询域的存在。
- 评估通过查看线程对齐覆盖"冠状病毒"。列和检查对齐。如果顶级路线的覆盖率低,局限于只有一长段的查询序列查询蛋白质或缺席的小区域,然后查询的蛋白质通常包含多个域和建议分裂序列和模型单独的域(图3)。
- 点击下载"下载的PDB格式的序列结构调整文件对齐"链接。这些对齐文件可以打开,在材料一节中列出的任何分子可视化方案,也可以用在结构建模(步骤1.4)中加入额外的限制。
6。在临时区议会的结构类似物
- 查看下表结果页(图4)结构对齐计划确定的十大结构类似物的第一个预测模型,为确定商标对齐20。一个TM -> 0.5分,显示检测到的模拟和模型,也有类似的拓扑结构,可用于确定查询蛋白16家族的结构类/蛋白质,而与TM -分数<0.3意味着一个随机结构的相似性。
- 分析序列的身份,并在区域结构对齐的RMSD"IDEN一个'和'的RMSD了 "的列,以评估模型的空间图案保护和结构模拟。目视检查对齐的颜色和对齐的残留物对,以确定这些结构上的保守残基和图案。
- 点击"临时区议会创"专栏访问RCSB网站,了解更多的PDB代码结构分类(SCOP,导管和PFAM)和职能部门的信息(EC编号,相关的GO术语和约束的配体)。
7。使用辅助因子的功能预测
- 在结果页面滚动分析查询的蛋白质的功能解释。蛋白质的功能是在三个背景下表列举,显示:酶委员会(EC)号码,基因本体论(GO)的条款,与配体结合位点。
- 查看"以旧换新得分"的RMSD,"A","IDEN一个 "和"冠状病毒"。在每个表中的列全球结构的相似性和保护模型和确定功能同源物(模板)之间的空间格局分析参数。
8。酶委员会人数预测
- 查看查询蛋白质的五大潜在酶在显示同源性"预测EC编号"表(图5)。置信水平的EC编号使用这些模板的预测显示"EC -分数"列。基于对benchmarking 23分析,查询和模板蛋白之间的功能相似(EC编号的第3位)能够可靠地使用EC -评分> 1.1解释。
- 查找功能的共识(EC编号)之间的模板,查询蛋白类似倍(即TM得分> 0.5)。如果有多个模板相同的EC编号和EC -> 1.1分,预测的置信水平是很高的。但是,如果欧共体得分高,但有一个缺乏确定命中的共识,然后预测变得不那么可靠和用户建议征询GO的长期预测。
- 点击EC编号为访问ExPASy酶数据库和分析功能的链接,包括详细的模板蛋白的催化反应,共同因素的要求和代谢途径。
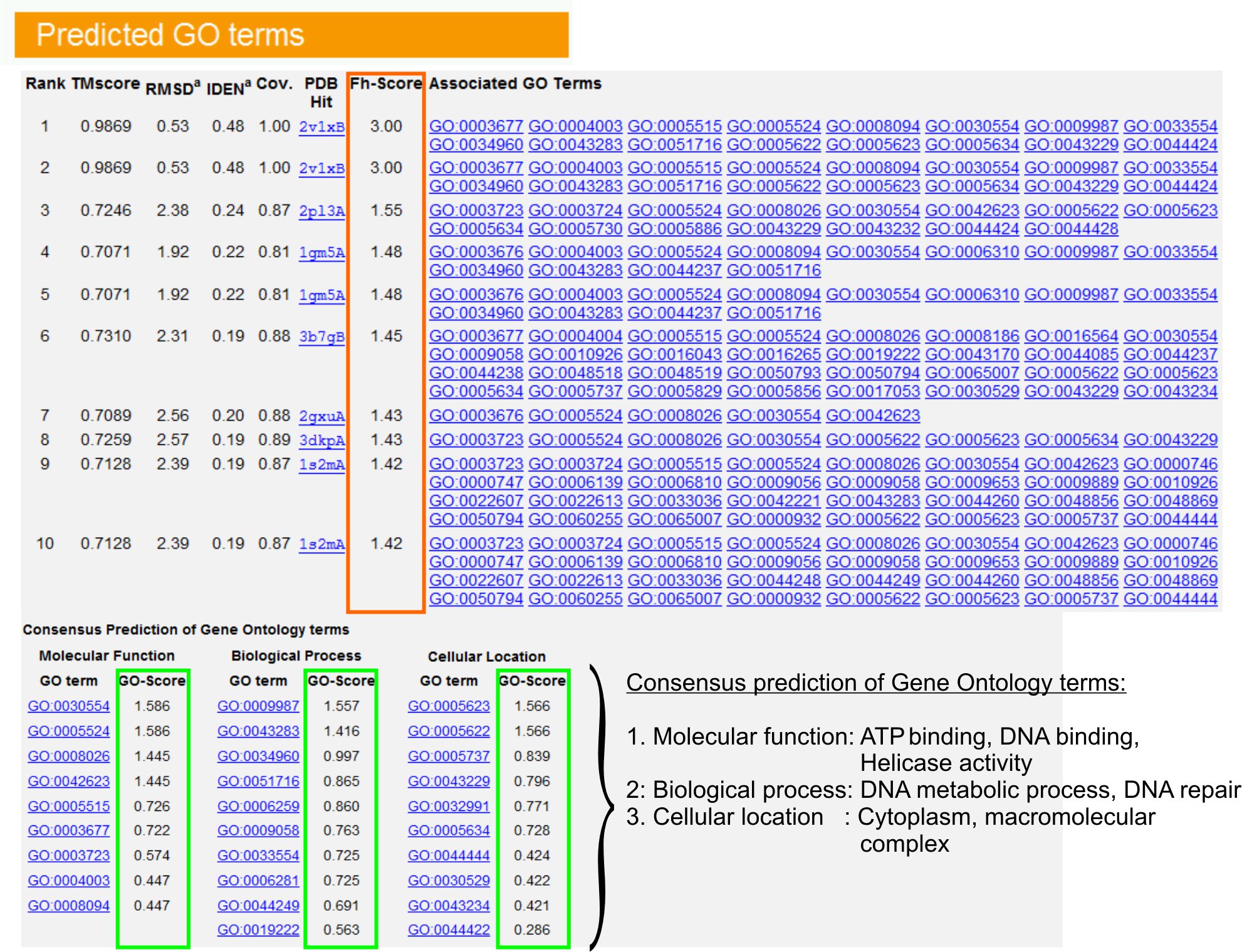
9。基因本体论(GO)的长期预测
- 查看"预言GO术语"表(图图6),以确定十大查询蛋白同系物在PDB库中,基因本体论(GO)的条款注明。每个蛋白质通常与多个GO术语,描述了其分子功能(MF)(BP)的生物过程和细胞组分(CC)的。每学期,点击访问朋友的网站,并分析其定义和血统。
- 分析FH -得分(功能的同源性得分)列访问之间的查询和模板蛋白的功能相似,估计转移这些蛋白质功能注释的信心水平。在我们的基准研究 23,50%的原生的GO术语可以使用一个56%的整体精度为0.8 FH得分截止,从首次发现模板正确识别。
- GO术语的"共识预测"表分析的功能模板之间的同意。这些常见的功能是用于预测查询(MF,BP和CC的GO术语)蛋白质和评估GO长期预测的置信水平(GO分数)。基于基准测试23,获得最好的假阳性和假阴性率预测GO得分截止= 0.5,减少在更深层次的本体水平预测的报道。
10。蛋白质 - 配体结合位点预测
- 向下滚动页面,以查看十大查询蛋白的配体结合位点预测的底部。预测的结合位点的基础上预测配体的构象,有着共同的结合口袋的数量排名。确定的最佳的结合位点已经显示在Jmol的applet。点击单选按钮来分析,预测和可视化的配体相互作用的残留。
- 分析评估模型和模板的结合位点之间的局部相似的BS得分列。基于基准9,BS得分> 1.1表示高的顺序和结构的SIMilarity附近的结合位点的预测模型和模板中的已知结合位点。
- 点击"下载"链接下载复杂的PDB格式的结构文件。用户可以打开这些文件中的任何分子可视化程序和交互式查看预测的结合位点和配体 - 蛋白质相互作用,在本地计算机上。
11。代表性的成果

图1我TASSER结果页的摘录显示(一)FASTA格式的查询序列;(B)二级结构预测和相关的信心分数;(三)溶剂可预测的残留物。在青色和红色矩形,分别突出分析的核心区域和潜在的水化网站查询。

图2。

图3:我TASSER结果页显示LOMETS 5线程方案十大确定的线程模板和路线的一个例子。线程路线的质量评估的基础上归Z - score模型(以绿色突出显示),其中一个值> 1反映了自信的对齐。模板中的不结盟残留物中查询到相应的残留物是相同的,都是突出的颜色来表示的保守残基/ Motif的存在,而在最缺乏的对齐顶部的模板表示的查询中的蛋白质和未对齐的残留存在多个域,对应域连接器区域。点击这里查看全尺寸版本的图3 。
{kind=link}

图4显示十大确定的结构类似物和结构的路线,所确定的结果页的例子。TM -对齐20结构对齐程序。类似物的排名是基于TM -得分的结构对齐(以蓝色突出显示)。一个TM -评分> 0.5表示比较的两个结构有一个类似的拓扑结构,而TM -分数<0.3意味着两个随机结构之间的相似性。结构上保持一致的残留对基于其氨基酸财产的颜色,而未对齐的地区是由表示" - "突出。ove.com/files/ftp_upload/3259/3259fig4large.jpg">点击这里查看图4的全尺寸版本。

图5:我TASSER的结果显示在PDB库查询蛋白质的酶的同源性的一个例子。 EC号预测的置信水平是分析的基础上EC -得分(以绿色突出显示),EC得分> 1.1表示查询和模板蛋白之间的功能相似(EC编号的第3位数字相同)。

图6:我TASSER结果显示页的一个例子查询蛋白质的长期预测。 FH -得分(橙色矩形)的基础上,在基因本体模板库查询蛋白质功能的同源性排名。得分最高的命中从这些常见的功能特点是由来以产生吃的是查询蛋白的最终长期预测。预测的GO术语的质量估计的基础上GO得分(以绿色显示),其中一去得分> 0.5表示一个可靠的预测。点击这里查看图6全尺寸的版本。
{kind=link}

图7:我TASSER结果页显示十大蛋白配体结合位点的预测,使用的辅助因子9算法的一个例子。排名是根据预测的结合位点的查询,分享共同的结合口袋的预测配体的构象。 BS得分(以红色突出显示)是本地序列和结构之间的相似性的预测和模板的结合位点的测量和分析的结合位点的口袋的保护非常有用。
les/ftp_upload/3259/3259fig8.jpg"/>
图8。外部约束用于指定残渣残留接触/距离限制的文件的一个例子。

图9:我TASSER服务器指定模板蛋白的克制文件的例子。用户可以指定查询模板对齐(一)FASTA格式;或(B)3D格式。

图10。排除在我TASSER结构建模过程的模板的示例文件。第一列包含的被排除在外的模板蛋白质的PDB ID。第二列是用来指定将使用其他类似的模板,在模板库的序列标识截止。
Discussion
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
上述协议是一个使用我的TASSER服务器的结构和功能建模的一般准则。虽然,这种自动化程序非常适用于大多数的蛋白质,人类的干预往往有助于显著提高建模精度,尤其是蛋白质缺乏在PDB库的密切模板。用户可以干预在我TASSER建模在以下方面:(一)多结构域蛋白的分裂;(b)提供外部约束,以改善结构组装;及(c)消除在建模的模板。
分裂多结构域蛋白:
许多长期的蛋白质序列通常包含多个域,灵活的连接器的地区,这使得其结构鉴定难以用实验和计算技术拴。然而,作为域是独立折叠实体,并可以执行不同的分子功能;宜分裂长的多结构域蛋白和模型,每个域分开。建模领域单独将不仅加快了预测过程,但也增加了查询模板对齐的质量,更可靠的结构和功能预测。
可以预见,在蛋白质序列域边界,使用免费提供的外部网上课程,如NCBI 的 CDD 24 PFAM 25或26 InterProScan。此外,如果LOMETS线程路线可供查询的蛋白,可域边界位于视觉识别很长一段未对齐的残留在顶部的线程模板(见第5.4步)。这些未对齐的地区,大多对应域连接器区域。如果多域模板已经在模板中的所有查询对齐域的PDB库,然后查询蛋白可以仿照全长。
提供外部约束
在我的TASSER结构装配模拟,主要是从线程模板LOMETS收集的空间限制的指导。有良好的线程命中(Norm. Z - score模型> 1)在模板库查询蛋白质,派生的空间限制,大多精度高,我TASSER将这些蛋白质生成高分辨率的结构模型。相反,查询的蛋白质,有弱或没有线程命中(Norm. Z分数 <1),收集了空间的限制,往往含有错误,因为模板对齐的不确定性。对于这些蛋白质的目标,用户指定的空间信息,可以提高预测模型的质量是非常有益的。用户可以提供两种方式我TASSER服务器的外部约束:A.指定联系人/距离限制
实验特点的跨残留的接触/距离,例如从核磁共振交叉连接实验,可以通过上传约束文件指定。一个例子文件是在图8所示,其中第1列指定类型的约束,即"距离"或"联系"。对于距离克制(测距),2和第4列含有残留物的位置(I,J),3和第5列中的残留物和第6列中包含的原子类型指定两个指定的原子之间的距离。对于接触限制(接触),2和第3列包含的残留量应在接触的位置(I,J)。这些联系的残留物对侧链中心之间的距离决定的基础上已知的PDB结构的观察距离。我TASSER将尝试绘制这些原子对指定的距离在结构细化模拟。
B.指定蛋白质的结构模板
LOMETS线程程序使用有代表性的PDB库,寻找可能的查询PROT褶皱EIN。虽然使用有代表性的结构库,有助于减少计算的序列结构路线所需的时间,它是可能错过一个很好的模板蛋白是在图书馆或模板可能没有被LOMETS线程方案确定,即使它是目前在库中。在这种情况下,用户应指定为模板所需的蛋白质的结构。
要指定一个额外的模板的蛋白质结构,用户可以上传PDB格式的结构文件,或指定在PDB库中存放的蛋白质结构的PDB ID。我的TASSER将生成的查询模板对齐, 使用鼓起方案23,将收集指定用户的模板和LOMETS模板,引导结构装配仿真空间的限制。由于LOMETS限制的准确性是针对不同的目标不同,LOMETS限制重量是容易(同源)TArgets比在硬盘(非同源)的目标,这已经在我们的基准培训系统调整。
用户还可以指定自己的查询模板路线。服务器接受对齐两种格式:FASTA格式(图9A)和3D格式(图9b)。 FASTA格式的标准和描述http://zhanglab。 ccmb.med.umich.edu / FASTA / 。 3D格式是类似的标准PDB格式( http://www.wwpdb.org/documentation/format32/sect9.html ),但来自模板的两个附加列被添加到原子记录(见图9B):
列1-30:原子(C -α)和残留查询序列的名称。
列31-54:从模板中的相应原子复制查询的C -α的原子坐标。
列55-59:在相应的模板残留数量的基础上对齐
列60-64:通讯残留在模板名称
排除模板蛋白质
蛋白质是灵活的分子,可以采取多种构象状态,以改变其生物活性。例如,许多蛋白激酶和膜蛋白的结构已经解决了active 和 inactive的构象。约束配体的存在或缺乏可造成大的构造运动。虽然所有的构象状态的模板是线程程序一样,它是可取的模式查询使用模板,只在一个特定的状态。在服务器上的一个新的选项允许用户排除在结构造型的模板蛋白。此功能也允许用户选择的造型模板的同源性水平。用户可以排除模板蛋白FROM我TASSER库:
A.指定序列的身份截止
用户可以使用这个选项排除我TASSER模板库的同源蛋白。同源性级别设置基于序列的身份截止,即除以查询顺序序列的长度之间的查询和模板蛋白相同的残渣的数量。例如,如果用户的类型,在"70%",在所提供的形式,所有模板蛋白质> 70%,我将我TASSER模板库中排除查询蛋白质序列的身份。
B.排除特定的模板蛋白
上传一个列表,其中包含要排除的结构的PDB的ID,可以排除特定的模板蛋白的I - TASSER模板库。图10显示了一个例子文件。由于相同的蛋白质可以存在多个条目,在PDB库,我TASSER SErver默认情况下,将排除指定的模板(Column1中)以及从库中,有一个身份的所有其他模板> 90%,到指定的模板。用户还可以指定一个不同的身份截止,如70%,所有与身份模板> 70%,到指定的模板蛋白将被排除在外。
Disclosures
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
没有利益冲突的声明。
Acknowledgements
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
该项目是支持部分由Alfred P. Sloan基金会NSF事业奖(DBI 1027394),和国家普通医学科学研究所(GM083107,GM084222)。
Materials
- FASTA格式的氨基酸序列为蓝本的蛋白质( 见 http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml) 。
- 与个人电脑接入互联网和Web浏览器。
- 分子可视化软件,如RASMOL或PYMOL,分析预测的三级结构和功能的网站。
References
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request Permission