

Summary
La formation de faisceaux d’actomyosine in vitro et la mesure de la génération de force d’ensemble de myosine à l’aide de pinces optiques sont présentées et discutées.
Abstract
Les myosines sont des protéines motrices qui hydrolysent l’ATP pour marcher le long des traces de filament d’actine (FA) et sont essentielles dans les processus cellulaires tels que la motilité et la contraction musculaire. Pour comprendre leurs mécanismes générateurs de force, la myosine II a été étudiée à la fois au niveau de la molécule unique (SM) et en tant qu’équipes de moteurs in vitro en utilisant des méthodes biophysiques telles que le piégeage optique.
Ces études ont montré que le comportement générateur de force de myosine peut différer considérablement lorsqu’il passe du niveau d’une seule molécule dans un arrangement à trois billes à des groupes de moteurs travaillant ensemble sur une surface rigide de bille ou de glissement de couverture dans un arrangement de glissement. Cependant, ces constructions de dosage ne permettent pas d’évaluer la dynamique de groupe de la myosine dans la hiérarchie structurelle viscoélastique comme elles le feraient dans une cellule. Nous avons développé une méthode utilisant des pincettes optiques pour étudier la mécanique de la génération de force par les ensembles de myosine interagissant avec plusieurs filaments d’actine.
Ces faisceaux d’actomyosine facilitent l’investigation dans un environnement hiérarchique et conforme qui capture la communication motrice et la sortie de force d’ensemble. La nature personnalisable du test permet de modifier les conditions expérimentales pour comprendre comment les modifications apportées à l’ensemble de myosine, au faisceau de filaments d’actine ou à l’environnement environnant entraînent des sorties de force différentes.
Introduction
Les protéines motrices sont essentielles à la vie, convertissant l’énergie chimique en travail mécanique 1,2,3. Les moteurs de myosine interagissent avec les filaments d’actine en faisant des pas le long des filaments comme une piste, et la dynamique des réseaux actine-myosine effectue la contraction musculaire, la motilité cellulaire, l’anneau contractile pendant la cytocinèse et le mouvement de la cargaison à l’intérieur de la cellule, entre autres tâches essentielles 3,4,5,6,7,8 . Étant donné que les myosines ont tant de rôles essentiels, une défaillance de la fonctionnalité du réseau myosine-actine peut entraîner le développement de maladies, telles que des mutations dans la chaîne lourde de myosine qui provoquent une hypercontractilité cardiaque dans la cardiomyopathie hypertrophique (CMH)9,10,11,12,13,14 . Dans la contraction musculaire, les moteurs individuels de la myosine coopèrent les uns avec les autres en travaillant comme un ensemble pour fournir l’énergie mécanique requise qui effectue le glissement relatif des AF 4,15,16,17,18. Les moteurs de myosine forment des ponts croisés entre les FA et utilisent les changements conformationnels dus à son cycle mécanochimique pour se déplacer collectivement vers l’extrémité barbelée des filaments alignés 17,18,19,20,21.
La mise au point d’essais quantitatifs de motilité in vitro au niveau SM à l’aide de techniques telles que le piégeage optique a facilité la collecte de détails sans précédent sur le fonctionnement des moteurs individuels de la myosine, y compris la mesure de la génération de force SM et de la taille des pas 22,23,24,25,26,27,28,29,30 . Finer et al. ont mis au point le test de piégeage optique « à trois billes » ou « haltères » pour sonder la mécanique de génération de force des moteurs à myosine II simples23,31. Comme la myosine musculaire II fonctionne en équipe pour contracter les FA mais n’est pas processive au niveau SM, l’orientation du test de piégeage optique a dû être réorganisée à partir de l’approche classique des perles liées au moteur32. Pour former le test d’haltères, deux pièges optiques ont été utilisés pour maintenir une mise au point automatique au-dessus d’un moteur de myosine lié à une perle fixée par un couvercle, et la force de sortie par le moteur unique a été mesurée par les mouvements de la mise au point automatique à l’intérieur du piège23.
Cependant, les forces SM et l’utilisation d’une orientation d’essai à un seul moteur/filament unique ne donnent pas une image complète de la génération de force au niveau du système, car de nombreuses protéines motrices, y compris la myosine II, ne fonctionnent pas isolément et ne fonctionnent souvent pas comme une somme de leurs parties 15,16,17,32,33,34,35,36 . Des structures plus complexes comprenant plus d’un moteur interagissant avec plus d’un filament sont nécessaires pour mieux comprendre la synergie des réseaux de myosine et d’actine 15,32. L’orientation du test d’haltères a été exploitée pour étudier la génération de force de petit ensemble en ayant plusieurs myosines attachées à une bille ou en utilisant un filament épais de myosine attaché à une surface et en permettant aux moteurs d’interagir avec la suspension AF 4,23,34,37,38,39,40.
D’autres essais de petit ensemble comprennent un essai de glissement filamentaire in vitro dans lequel les moteurs de myosine sont recouverts sur une surface de glissement de couverture, et une perle liée à une AF est utilisée pour sonder la force générée par l’équipe de moteurs 4,35,36,38,39,40,41,42,43 . Dans ces deux cas, les myosines sont liées à une surface rigide – perle ou lamelle de couverture – et utilisent une AF. Dans ces cas, les moteurs ne sont pas capables de se déplacer librement ou de communiquer entre eux, et le fait d’avoir des myosines rigidement liées ne reflète pas l’environnement hiérarchique conforme dans lequel les moteurs travailleraient ensemble dans le sarcomère32. Des études antérieures ont suggéré que la myosine II peut détecter son environnement et s’adapter en conséquence aux conditions changeantes de concentration viscoélastique ou motrice en modifiant des caractéristiques telles que la génération de force et le rapport de service41,44,45. Il est donc nécessaire de mettre au point un test de piégeage optique qui favorise et capture la communication motrice et la conformité du système afin de brosser un tableau plus réaliste des fondements mécanistes de la génération de force d’ensemble de la myosine II.
Ici, nous avons développé une méthode pour coupler la structure hiérarchique in vitro avec le piégeage optique en formant des faisceaux d’actomyosine ou des sandwichs constitués de plusieurs moteurs de myosine interagissant entre deux filaments d’actine. Cette géométrie de dosage modulaire a la capacité de sonder directement comment les facteurs moléculaires et environnementaux influencent la génération de la force de myosine d’ensemble. De plus, l’étude des mécanismes de génération de force à travers ces ensembles actine-myosine a le potentiel d’aider à modéliser et à comprendre comment les tâches cellulaires à grande échelle, telles que la contraction musculaire, se propagent à partir du niveau moléculaire 9,10,13.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Graver les lamelles de couverture
- Dissoudre 100 g de KOH dans 300 mL d’éthanol à 100 % dans un bécher de 1 000 mL. Remuer avec une barre d’agitation jusqu’à ce que la majorité du KOH soit dissoute.
ATTENTION: La solution concentrée de KOH peut causer des brûlures et endommager les vêtements. Portez des gants, une protection oculaire et une blouse de laboratoire. - Placez les lamelles de couverture individuellement dans des supports de nettoyage de lames de couverture.
REMARQUE: Les racks sont conçus avec des fentes qui maintiennent des lamelles de recouvrement simples espacées pour permettre la gravure et le rinçage sur chaque face de la lamelle de couverture, des trous de vidange dans le fond et faites d’un matériau pouvant résister aux conditions de gravure difficiles. Ils peuvent être fabriqués sur mesure ou achetés dans le commerce. - Préparez et étiquetez trois béchers de 1 000 ml : un avec 300 ml d’éthanol et deux béchers avec 300 ml d’eau osmosée (osmose).
REMARQUE: Ici, l’eau osmosée provient d’un purificateur d’eau de laboratoire, mais elle peut également être achetée dans le commerce si un purificateur local n’est pas disponible. - Placez chacun des quatre béchers dans un sonicateur de bain pour dégazer pendant 5 min.
- Immerger un rack de lamelles de couverture dans le bécher de KOH et d’éthanol et soniquer pendant 5 min.
- Transférer le rack de lamelles de couverture du bécher KOH/éthanol au bécher contenant uniquement de l’éthanol. Trempez la grille de haut en bas dans le bécher jusqu’à ce qu’il n’y ait plus de perlage.
REMARQUE: Veillez à ne pas déranger les lamelles de couverture ou à ne pas laisser tomber de force le rack dans le bécher. Cela entraînera la sortie des lamelles de recouvrement du rack ou provoquera des éclaboussures chimiques. - Transférez délicatement le rack de lamelles de couverture du bécher à éthanol dans un bécher d’eau, en plongeant de haut en bas jusqu’à ce qu’il n’y ait plus de perlage.
- Plongez le rack de lamelles de couverture dans le bécher d’eau qui n’a pas encore été utilisé et soniquez à nouveau pendant 5 min.
- Utilisez une bouteille pour asperger le rack de lamelles de couverture avec de l’eau jusqu’à ce qu’elle s’écoule doucement des lamelles de couverture. Répétez avec l’éthanol.
- Placer les grilles à sécher dans un four à 90 °C pendant 20 min. Entreposer les étagères des lamelles de recouvrement gravées à température ambiante dans des contenants fermés pour éviter toute contamination avant utilisation.
2. Polymérisation du filament d’actine
- Créer une solution T
- Dans un tube conique de 50 mL, ajouter 3,94 g de Tris-HCl et 0,147 g de CaCl2. Ajouter de l’eau osmosée pour obtenir un volume total de 50 ml et bien mélanger.
NOTA: Les concentrations finales de la solution Tsont respectivement de 500 mM de Tris-HCl et de 20 mM de CaCl2. - Étiqueter le tube Solution T et le conserver à 4 °C.
- Dans un tube conique de 50 mL, ajouter 3,94 g de Tris-HCl et 0,147 g de CaCl2. Ajouter de l’eau osmosée pour obtenir un volume total de 50 ml et bien mélanger.
- Créer un tampon TC
- Mélanger 40 mL d’eau d’osmose inverse et 1,5 mL de solution T dans un tube conique de 50 mL. Changez le pH à 8,0 en ajoutant de petites quantités de KOH concentré. Ajouter de l’eau pour obtenir 50 ml de la solution et vérifier le pH. Ajustez le pH si nécessaire.
REMARQUE : Le tampon TC final contient 5 mM de Tris-HCl et 0,2 mM de CaCl2à pH 8. - Étiqueter le tube TC et le conserver à 4 °C.
- Mélanger 40 mL d’eau d’osmose inverse et 1,5 mL de solution T dans un tube conique de 50 mL. Changez le pH à 8,0 en ajoutant de petites quantités de KOH concentré. Ajouter de l’eau pour obtenir 50 ml de la solution et vérifier le pH. Ajustez le pH si nécessaire.
- Créer un tampon FC
- Ajouter 85 mL d’eau d’OI, 10 mL de Solution T, 3,73 g de KCl et 0,041 g de MgCl2 dans un flacon tampon de 100 mL. Modifier le pH à 7,5 en ajoutant de petits volumes de KOH concentré. Ajouter de l’eau pour obtenir un volume final de 100 mL et vérifier le pH.
REMARQUE : Le tampon FC final contient 500 mM de Tris-HCl, 500 mM de KCl, 2 mM de MgCl 2 et 2 mM de CaCl2 à pH 7,5. - Étiqueter le tube FC et le conserver à 4 °C.
- Ajouter 85 mL d’eau d’OI, 10 mL de Solution T, 3,73 g de KCl et 0,041 g de MgCl2 dans un flacon tampon de 100 mL. Modifier le pH à 7,5 en ajoutant de petits volumes de KOH concentré. Ajouter de l’eau pour obtenir un volume final de 100 mL et vérifier le pH.
- Préparer le tampon général d’actine (GAB).
- Mélanger 485 μL de tampon TC, 10 μL d’ATP 10 mM et 5 μL de DTT 50 mM dans un tube microcentrifugeuse.
REMARQUE : Les conditions tampons finales sont de 5 mM Tris-HCl, 0,2 mM CaCl 2, 0,5 mM TNT et0,2 mM ATP. - Étiquetez-le comme GAB et conservez-le à 4 °C.
- Mélanger 485 μL de tampon TC, 10 μL d’ATP 10 mM et 5 μL de DTT 50 mM dans un tube microcentrifugeuse.
- Préparer le tampon de polymérisation de l’actine (APB).
- Mélanger 455 μL de tampon FC, 25 μL d’ATP 100 mM et 20 μL de DTT 50 mM dans un tube microcentrifugeux.
NOTE: Les conditions tampons finales sont 50 mM Tris-HCl, 500 mM KCl, 2 mM MgCl 2, 2 mM CaCl 22 mM TNT et 5 mM ATP. - Étiquetez le tube comme APB et conservez-le à 4 °C.
- Mélanger 455 μL de tampon FC, 25 μL d’ATP 100 mM et 20 μL de DTT 50 mM dans un tube microcentrifugeux.
- Reconstituer l’actine
- Reconstituer l’actine des muscles squelettiques de lapin en ajoutant 100 μL d’eau désionisée à un flacon de 1 mg d’actine lyophilisée. Bien mélanger en pipitant doucement de haut en bas. Aliquote dans des échantillons de 5 μL, surgeler et conserver les aliquotes d’actine à 10 mg/mL à -80 °C.
- Reconstituer l’actine des muscles squelettiques de lapin biotinylés en ajoutant 20 μL d’eau osmosée. Aliquote dans des échantillons de 5 μL, surgeler et conserver les aliquotes d’actine biotinylée de 1 mg/mL à -80 °C.
- Polymérisation d’actine non marquée avec stabilisation de la phalloïdine rhodamine
- Décongeler un flacon de 10 mg/mL d’actine et le conserver sur la glace.
- Préparer un tampon GAB frais, ajouter 100 μL de GAB à l’aliquote d’actine et mélanger en pipitant doucement de haut en bas. Incuber la solution sur glace pendant 1 h.
- Préparer l’APB frais pendant l’incubation. Après incubation, polymériser l’actine en filaments en ajoutant 11 μL d’APB à la solution d’actine. Bien mélanger en pipitant doucement de haut en bas. Placer sur la glace pendant 20 min.
- Ajouter 5 μL de phalloïdine marquée à la rhodamine à la solution de filament d’actine fraîchement polymérisée. Laisser sur la glace dans l’obscurité pendant 1 h.
- Conservez le flacon d’actine de rhodamine enveloppé dans du papier d’aluminium dans l’obscurité à 4 °C.
REMARQUE: Il est suggéré d’utiliser ces filaments pendant une période maximale de 1 semaine. La qualité de l’AF peut être confirmée chaque jour grâce à une imagerie rapide d’une cellule d’écoulement contenant uniquement des AF et à la visualisation quotidienne de filaments cohérents.
- Polymérisation d’actine biotinylée avec stabilisation de la phalloïdine Alexa Fluor 488
- Décongeler un flacon de 10 mg/mL d’actine et 1 flacon de 1 mg/mL d’actine biotinylée et les conserver sur la glace.
- Faire un tampon GAB frais.
- Combiner les deux flacons (étape 2.8.1) dans un rapport actine:actine biotinylée de 10:1. Ajouter 100 μL de GAB au mélange d’actine et bien mélanger en pipetant doucement de haut en bas. Incuber sur glace pendant 1 h.
- Faire de l’APB frais pendant l’incubation.
- Après l’étape d’incubation, polymériser l’actine en ajoutant 11 μL d’APB à la solution d’actine. Bien mélanger en picoretant doucement de haut en bas. Incuber sur glace pendant 20 min.
- Ajouter 5 μL de phalloïdine marquée Alexa Fluor 488 et incuber sur de la glace dans l’obscurité pendant 1 h.
- Conservez le flacon d’actine biotinylée enveloppé dans du papier d’aluminium dans l’obscurité à 4 °C.
NOTE: Ces filaments peuvent être utilisés pendant une période maximale de 1 semaine.
3. Préparation de la myosine et des billes
- Reconstituer la myosine II
- Faire tourner brièvement vers le bas (~5 s) la myosine II squelettique lyophilisée pour la recueillir au fond du tube à l’aide d’une minicentrifugeuse standard.
- Reconstituer la myosine à 10 mg/mL en ajoutant 100 μL de DTT 1 mM préparé dans de l’eau d’OI.
- Diluer la solution mère de myosine 10x en ajoutant 10 μL de myosine à 10 mg/mL à 90 μL de DTT 1 mM dans de l’eau d’osmose inverse. Fabriquer des aliquotes de petit volume (1 à 5 μL), les congeler et les conserver à -80 °C.
NOTE: L’activité de la myosine peut être confirmée en effectuant un test de filament de glissement standard tel que publié précédemment46,47. Voir la discussion pour une brève description.
- Nettoyage des billes enrobées de streptavidine
- Diluer 20 μL de billes de streptavidine de 1 μm dans 80 μL d’eau osmosée. Laver quatre fois en essoretant à 9 600 × g et en se reconstituant dans 100 μL d’eau osmosée.
- Sonicer pendant 2 min à 40% d’amplitude et conserver les billes lavées sur un rotateur à 4 °C.
4. Préparation des cellules d’écoulement
- Préparer une solution de poly-l-lysine (LPL) en ajoutant 30 mL d’éthanol à 100 % dans un tube de 50 mL et en ajoutant 200 μL de poly-l-lysine à 0,1 % p/v dans de l’eau et bien mélanger.
- Ajouter une lamelle de couverture gravée à la solution PLL et laisser tremper pendant 15 min. Retirez le bordereau à l’aide d’une pince à épiler, en prenant soin de ne toucher que le bord du couvercle lorsqu’il est tiré vers le haut du tube (voir Figure 1A-C). Attrapez les bordereaux par les bords avec une main gantée.
- Sécher le bordereau de couverture avec une compagnie aérienne filtrée jusqu’à ce qu’il ne reste plus d’éthanol et aucun résidu sur le couvercle.
- Appliquez deux morceaux de ruban adhésif double face au milieu d’une lame de microscope, à 3-4 mm l’un de l’autre. Déchirez ou coupez l’excès de ruban adhésif qui pend du bord de la glissière.
- Ajouter la lame de couverture enduite de PLL sur le dessus de la bande perpendiculairement à l’axe long de la lame de microscope (formant un T) pour former un canal.
- Utilisez un petit tube pour comprimer la lamelle de couverture sur le ruban et la lame de microscope à fond jusqu’à ce que la bande soit transparente (Figure 1A). Assurez-vous qu’il n’y a pas de bulles dans la bande, car cela peut provoquer une fuite du canal d’écoulement.
REMARQUE: La cellule d’écoulement peut contenir un volume de 10-15 μL.
5. Préparation du faisceau d’actomyosine
- Dans des tubes séparés, diluer chaque type de filament d’actine (marqué à la rhodamine et au 488 biotinylé) 600x en mélangeant 0,5 μL d’actine marquée respective avec 300 μL d’APB. Ajouter 5 μL supplémentaires de la phalloïdine étiquetée correspondantement dans chaque tube et incuber sur de la glace dans l’obscurité pendant 15 minutes.
- À la solution d’actine biotinylée, ajouter un système de piégeage de l’oxygène de 1 μL de bêta-D-glucose à 500 mg/mL, de 1 μL de glucose oxydase à 25 mg/mL et de 1 μL de catalase à 500 unités/mL. Ajouter 1 μL d’ATP 100 mM et 1 μL de billes de streptavidine diluées et nettoyées 100x. Remuer doucement avec un embout de pipette. Placez la suspension sur un rotateur à 4 °C pendant que le reste du faisceau d’actomyosine est en cours d’assemblage.
- Ajouter 15 μL d’actine rhodamine diluée à la cellule d’écoulement de la LPL (Figure 1D). Aspirez l’excès de solution à travers la cellule d’écoulement, mais ne laissez pas le canal d’écoulement s’assécher. Incuber pendant 10 min dans une chambre d’humidité.
REMARQUE: Les chambres d’humidité peuvent être fabriquées à partir de boîtes d’embout de pipette vides avec de l’eau ajoutée au fond et le couvercle recouvert de papier d’aluminium pour bloquer la lumière. - Préparer une solution de caséine à 1 mg/mL dans de l’APB.
- Ajouter 15 μL de caséine à 1 mg/mL pour éviter la liaison non spécifique des composants suivants (figure 1E). Incuber pendant 5 min dans une chambre d’humidité.
- Ajouter la concentration désirée de myosine à l’actine biotinylée et à la suspension de billes de l’étape 5.2. Remuer doucement avec l’embout de la pipette, puis ajouter immédiatement 15 μL de la suspension de l’étape 5.2 + la concentration de myosine souhaitée dans la cellule d’écoulement (Figure 1F,G). Incuber pendant 20 min. Scellez les extrémités ouvertes de la cellule d’écoulement avec du vernis à ongles pour éviter l’évaporation pendant les expériences d’imagerie et de piégeage optique.
REMARQUE : Une concentration de 1 μM en solution de myosine donne un regroupement robuste et peut être utilisée comme point de départ pour la personnalisation souhaitée du dosage (voir la figure 2).
6. Mesures de force à l’aide d’un piège optique (NT2 Nanotracker2)
REMARQUE: Bien que le protocole ci-dessous soit spécifiquement destiné au système NT2, ce test peut être utilisé avec d’autres systèmes de piégeage optique, y compris ceux qui sont construits sur mesure, qui ont également des capacités de fluorescence. Le flux de travail général reste le même pour obtenir la surface de la diapositive en évidence, effectuer des étalonnages de cordons et acquérir des données en trouvant des faisceaux d’actine fluorescents. Pour le système NT2, la figure supplémentaire S1, la figure supplémentaire S2, la figure supplémentaire S3, la figure supplémentaire S4, la figure supplémentaire S5, la figure supplémentaire S6 et la figure supplémentaire S7 fournissent des détails sur le système de piégeage optique et l’interface logicielle.
- Allumez le boîtier de commande et le laser (Figure supplémentaire S1).
- Démarrez le logiciel informatique du piège optique en cliquant sur l’icône JPK Nanotracker sur le bureau.
- Réveillez la radiocommande en cliquant sur le bouton Logitech au centre (Figure supplémentaire S2).
- Allumez le module de fluorescence en activant l’interrupteur marche/arrêt (Figure supplémentaire S3).
- Tournez la tourelle du cube filtrant pour l’imagerie en fond clair (figure supplémentaire S4).
- Une fois le système prêt, allumez le laser à l’aide du bouton d’alimentation laser situé dans le coin inférieur gauche de l’écran à 50 mW et laissez-le se stabiliser pendant 30 min (figure supplémentaire S5).
- Cliquez séquentiellement sur les boutons Illumination, Caméra, Objectif et Mouvement de scène dans le logiciel pour afficher ces fenêtres de visualisation et de manipulation pendant l’expérience. Allumez l’éclairage du microscope en cliquant sur le bouton On/Off et réglez-le sur la puissance maximale en cliquant et en faisant glisser la barre vers la droite (Figure supplémentaire S5).
- Ouvrez la zone d’échantillon et retirez le porte-échantillon de l’étage du microscope. Ajoutez la cellule d’écoulement, fixez-la avec les porte-échantillons métalliques et assurez-vous que la lame avec le bordereau de couverture est en bas.
- Ajouter 30 μL d’eau osmosée au centre de l’objectif inférieur. Ne laissez pas l’embout de la pipette toucher l’objectif. Réinsérez l’étape d’échantillonnage.
REMARQUE : Comme le système NT2 utilise un objectif d’immersion dans l’eau comme objectif de piégeage, le milieu d’immersion peut être différent en fonction de l’objectif de piégeage dans la configuration de l’utilisateur. - Soulevez l’objectif inférieur à l’aide des flèches de commande à l’écran ou L2 de la télécommande jusqu’à ce que le cordon d’eau touche la lamelle de couverture (figure supplémentaire S5).
- Abaissez l’objectif supérieur jusqu’à ce qu’environ la moitié de la distance jusqu’à la cellule d’écoulement soit atteinte à l’aide des flèches à l’écran ou R2 de la télécommande. Ajouter 170 μL d’eau osmosée au sommet de la cellule d’écoulement directement sous l’objectif supérieur. Abaisser l’objectif supérieur jusqu’à ce qu’il brise la tension superficielle de l’eau et forme un ménisque.
- Déplacez l’étage du microscope à l’aide du patin fléché de la télécommande jusqu’à ce que le bord de la bande adjacent au canal d’écoulement soit atteint. Fermez la porte de l’échantillon.
REMARQUE: Un « clic » à la fermeture de la porte de l’échantillon indique que l’obturateur laser est maintenant ouvert. Il s’agit d’un dispositif de sécurité qui ne permet au volet de s’ouvrir que si la porte est fermée. - À l’aide de la fenêtre Objectif à l’écran, mettez le bord de la bande au point en amenant l’objectif inférieur nommé Objectif laser vers le haut en cliquant sur la flèche supérieure à l’aide des commandes à l’écran. Faites de même pour l’objectif supérieur en cliquant sur la flèche du bas (Figure supplémentaire S5).
REMARQUE: Les flèches doubles déplacent l’objectif ou la scène plus rapidement. Le bord de la bande est utilisé pour la mise au point car il s’agit d’un objet volumineux, facile à trouver, proche de la surface de la lamelle de couverture. Les bulles d’air à l’intérieur de la bande sont une autre option. Cependant, cela n’est pas nécessaire si l’utilisateur dispose d’une routine automatisée pour trouver la mise au point de surface ou d’une méthode interne préférée. - Une fois la bande mise au point, fermez partiellement le diaphragme en haut du piège optique. Abaissez l’objectif supérieur jusqu’à ce que la forme polygonale de l’iris soit visible. Mettez ces bords au point, rouvrez l’iris, puis associez les objectifs en cliquant sur l’icône Cadenas (Figure supplémentaire S5).
- Trouvez un cordon flottant et piégez-le en cliquant sur le bouton Trap Shutter, qui ouvrira l’obturateur et permettra au laser de piégeage de frapper l’échantillon. Cliquez sur le curseur Interruption à l’écran et faites-le glisser pour déplacer l’emplacement du laser de recouvrement. Une fois piégé, calibrez le cordon pour corréler les mesures de tension à la force et au déplacement.
- Cliquez sur le bouton Calibration . Ajustez la routine d’étalonnage en fonction de l’analyse des spectres de puissance et ajustez la fréquence de coin dans le logiciel pour les directions X, Y et Z (figure supplémentaire S6).
- Cliquez sur Paramètres. Tapez le diamètre de la perle (1 000 nm) et tapez la température de l’étage située en bas à gauche de la fenêtre du logiciel. (voir la figure supplémentaire S6).
- Cliquez sur Piège 1. Cliquez sur X Signal. Cliquez sur Exécuter pour effectuer l’ajustement de la fréquence d’angle. Cliquez et faites glisser dans la fenêtre pour optimiser l’ajustement de la fonction. Cliquez sur Utiliser pour connaître les valeurs de sensibilité et de rigidité. Cliquez sur Accepter les valeurs. Répétez l’opération pour les signaux Y et Z. Fermez la fenêtre. (voir la figure supplémentaire S6).
REMARQUE : Les routines d’étalonnage du cordon sur d’autres systèmes de piégeage optique ou des systèmes sur mesure qui ont été rigoureusement testés par l’utilisateur, comme la méthode d’équipartition ou la méthode de force de traînée, sont également acceptables57,58. - Trouvez un faisceau d’actomyosine en recherchant les billes liées aux AF sur la surface de la lame.
- Lorsqu’un cordon non encombré par d’autres billes flottantes est détecté, observez les AF autour de lui par imagerie par fluorescence pour vérifier la présence d’un faisceau.
- Vérifiez qu’un faisceau est présent en recherchant les deux AF fluorescents colocalisés. Allumez la source de lumière blanche et utilisez le cube filtrant approprié pour imager chaque filament d’actine en tournant la tourelle (cubes de filtre d’excitation de 488 nm et 532 nm pour Alexa Fluor 488 et l’excitation à la rhodamine, respectivement). Voir la figure supplémentaire S4.
REMARQUE: Une expérience de contrôle pour vérifier l’intensité de fluorescence des AF simples peut être utile pour identifier les faisceaux composés d’un seul filament marqué à la rhodamine 488 et à la rhodamine, ou applicable à n’importe quel ensemble de fluorophores que l’utilisateur choisit d’utiliser. - Une fois vérifié, trapez le cordon attaché au filament supérieur du paquet en cliquant sur le bouton Trap Shutter.
- Utilisez les commandes à l’écran pour enregistrer les données en cliquant sur le bouton Oscilloscope (Figure supplémentaire S7). Pour visualiser les mesures sans enregistrer les données, cliquez sur Démarrer. Pour enregistrer toutes les données, cliquez sur Enregistrement automatique. Pour enregistrer les mesures, cliquez sur Démarrer l’enregistrement. Choisissez les données à visualiser en temps réel (position, force, direction x, direction y) en choisissant dans le menu déroulant le signal X ou le signal Y. N’oubliez pas que xdirection est de gauche à droite et que la direction y est de haut en bas sur l’écran. Voir la figure supplémentaire S7.
REMARQUE: Les données seront enregistrées sous forme de fichiers .out et incluent l’heure, la tension, le déplacement et la force pour chaque direction. Ces fichiers peuvent être exportés dans d’autres logiciels pour la visualisation et l’analyse.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Les cellules d’écoulement contenant les systèmes de faisceaux d’actomyosine sont de conception standard, constituées d’une lame de microscope et d’une lamelle de couverture gravée séparée par un canal constitué de ruban adhésif double face (Figure 1). Le test est ensuite construit à partir de la fiche de couverture en utilisant des introductions par étapes comme décrit dans le protocole. Le test final consiste en des filaments d’actine marqués à la rhodamine ; la concentration de myosine souhaitée (1 μM a été utilisé pour les résultats représentatifs de la figure 2 et de la figure 3); filaments d’actine biotinylés, marqués Alexa Fluor 488; billes de streptavidine de 1 μm; le système de récupération de l’oxygène; ATP; et tampon APB. Plusieurs faisceaux seront formés par cellule d’écoulement, et les concentrations d’actine décrites ci-dessus donnent un espacement adéquat entre les faisceaux pour assurer l’absence d’interactions indésirables. Cela facilite également l’obtention de plusieurs mesures de force par cellule d’écoulement pour augmenter l’efficacité de l’acquisition de données. Les profils de force doivent pouvoir être reproductibles à l’intérieur d’une cellule d’écoulement et d’une cellule d’écoulement à l’autre.
Bien que le protocole ci-dessus soit axé sur l’utilisation d’une installation de piégeage optique commerciale, la cellule d’écoulement et le test présentés ici pourraient facilement être utilisés pour un instrument commercial différent ou une installation de piégeage optique sur mesure couplée à un microscope ou à une étape de microscope et possédant des capacités d’imagerie par fluorescence. Une fois que tous les ajouts de cellules d’écoulement sont terminés conformément au protocole ci-dessus, les faisceaux d’actomyosine sur la lame (Figure 1) sont prêts pour une mesure immédiate. La cellule d’écoulement est ajoutée à l’étage du microscope à piège optique, plusieurs mesures d’étalonnage des billes sont acquises et les faisceaux sont identifiés par colocalisation en fluorescence des filaments du faisceau. Un cordon lié à un faisceau est piégé et le déplacement et la mesure de la force correspondante commencent. L’utilisateur peut observer l’acquisition de données en temps réel sur l’écran de l’ordinateur. Selon la concentration de myosine utilisée dans la cellule d’écoulement, le faisceau pourrait commencer à présenter un mouvement substantiel immédiatement, ou il peut prendre 30 s-1 min pour voir efficacement une augmentation du déplacement / force.
Une trace de force représentative est montrée à la figure 3A où les moteurs de myosine présentent une montée en force constante suivie d’un plateau. Il est typique de voir ces types de traces se développer sur 2-5 min. Cependant, il est également possible de mesurer des faisceaux d’actomyosine qui ne génèrent aucune force nette (Figure 3B). Ces traces apparaissent comme du bruit de base ou ne présentent pas d’augmentation nette substantielle de la force au-delà de 90 s. Cela est probablement dû à une faible concentration locale du moteur qui ne permet pas un glissement productif, ou le faisceau est dans une orientation parallèle défavorable où les extrémités plus et moins des filaments sont alignées.
Comme le contenu de la cellule d’écoulement peut être susceptible d’être dégradé par l’éclairage incident et le laser de piégeage, le chauffage local sur la lame au fil du temps et la génération d’espèces d’oxygène radicalaires, il est fortement conseillé de ne pas utiliser la même cellule d’écoulement pendant plus de 1 h. Pour une efficacité maximale, il est suggéré d’avoir un autre test en incubation tout en acquérant des données. La trace de déplacement/force peut être exportée du logiciel de piégeage optique vers Excel, Matlab, Igor ou d’autres programmes de gestion de données pour un filtrage et une analyse plus poussés. Les données qui peuvent être extraites de ces expériences d’ensemble/faisceau de piégeage optique comprennent différents types de profils de génération de force (ligne de base, rampe/plateau) dans des conditions d’essai variables, la vitesse de génération de force, la génération de force maximale, le comportement cinétique et pas d’ensemble à travers les tailles de pas et les temps de séjour entre les étapes ou les équipes d’étapes, ainsi que le rapport de service. L’utilisateur peut également modifier les conditions de dosage pour comparer comment l’ajout de différents types de moteurs de myosine, l’ajout de protéines de liaison à l’actine ou la modification des conditions tampons influencent ces caractéristiques de génération de force d’ensemble.
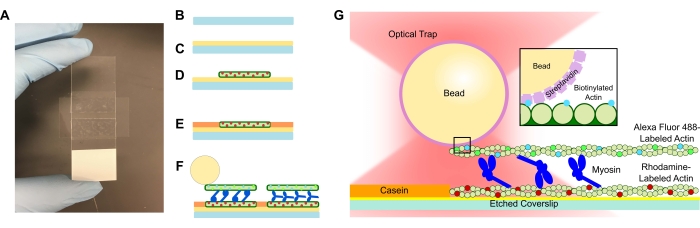
Figure 1 : Schéma du dosage. (A-C) Les lamelles de couverture gravées sont recouvertes de poly-L-lysine et utilisées pour former la cellule d’écoulement à l’aide d’un ruban adhésif double face et d’une lame de microscope. Les introductions chronométrées et les étapes d’incubation décrites dans le protocole aboutissent à une actine stabilisée à la phalloïdine marquée à la rhodamine comme matrice ou filament inférieur (D), suivie d’un blocage de la caséine pour empêcher la liaison non spécifique (E), et (F) à l’actine biotinylée stabilisée à la phalloïdine Alexa Fluor 488 comme cargaison ou filament supérieur, et à des équipes de myosine II qui séparent les filaments et génèrent de la force lorsque l’ATP est introduit. La géométrie des moteurs et la nature de la réticulation à l’intérieur du faisceau peuvent varier dans différentes conditions, telles que la concentration en sel59. Des études antérieures ont démontré que le domaine de la queue de la myosine a la capacité d’interagir avec les filaments d’actine et de ralentir la motilitéd’ensemble 46. Cependant, les têtes de myosine dans les expériences de méromyosine lourde démontrent la liaison de chaque tête aux filaments d’actine adjacents60. (G) Les billes de streptavidine sont utilisées comme poignée optique pour le piège et se lient uniquement au filament d’actine biotinylé de la cargaison, ce qui aide à valider que les faisceaux appropriés sont formés sur la lame. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 2 : Fibrés d’actomyosine fluorescents. Quatre rencontres différentes de filaments et de faisceaux d’actine dans le test de faisceaux présentés à la figure 1. Le filament d’actine biotinylé cargo supérieur avec le canal de phalloïdine Alexa Fluor 488 est montré à gauche, et le filament d’actine du bas avec le canal de phalloïdine rhodamine est à droite. En bas, la même figure est représentée avec des lignes colorées superposées pour aider à guider l’œil. (A) Un filament d’actine supérieur se trouve près d’un filament d’actine inférieur mais a une superposition incomplète. Cela ne serait pas utilisé pour les expériences de bundle. (B) Les filaments d’actine supérieur et inférieur sont colocalisés, et l’intensité de chaque filament confirme qu’il s’agit chacun de filaments uniques dans le faisceau. Ce serait un bon candidat pour les expériences de bundle. (C) Un gros paquet de filaments de rhodamine auto-assemblés se trouve sur le fond. Bien qu’il y ait un filament d’actine supérieur correspondant qui est colocalisé, il y a trop de filaments inférieurs présents; Ainsi, il ne serait pas utilisé pour des expériences de faisceaux. C’est aussi un exemple de la façon dont lorsque plusieurs filaments d’actine du même type sont groupés, l’intensité de fluorescence augmente. L’utilisateur peut l’utiliser comme jauge pour juger des filaments simples par rapport à des faisceaux du même type de filament. (D) Un filament inférieur est présent sans filament supérieur correspondant, ce qui confirme également l’absence de purge. Cela ne serait pas utilisé pour les expériences de bundle. Nous notons que l’intensité des filaments dans le canal Alexa Fluor 488 est faible et pensons que cela est dû au jeu de filtres utilisé (Filtre Set 09 de Zeiss). Le jeu de filtres utilisé pour le canal rhodamine est le jeu de filtres 43 de Zeiss. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 3 : Génération de force d’ensemble de la myosine II. Des traces représentatives de moteurs de myosine II squelettique générant une force dans la hiérarchie structurelle de l’actine in vitro construite. Les moteurs de la myosine travaillent ensemble pour générer collectivement et de manière productive de la force jusqu’à ce qu’un plateau soit atteint et que la force soit maintenue (A) ou subisse une antagonisation près de la ligne de base (B). Veuillez cliquer ici pour voir une version agrandie de cette figure.
Figure supplémentaire S1 : piège optique Bruker/JPK Nanotracker2. (A) Écran d’ordinateur. (B) Clavier d’ordinateur. (C) Tour d’ordinateur. (D) Boîtier contrôleur. (E) Alimentation laser. F) Boîtier optique à piège optique. (G) Microscope inversé. (H) Porte à l’étage du microscope. (I) Curseur polariseur pour basculer entre l’imagerie de contraste de fond clair et d’interférence différentielle. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S2 : Télécommande pour piège optique. (A) Clavier pour positionner la scène motorisée. (B-C) Ajustez la position du piège. (D) A, X et B allument et éteignent respectivement l’obturateur principal, l’obturateur 1 et l’obturateur 2. (E) Le bouton Logitech est utilisé pour réveiller la manette. (F) Les boutons haut et bas utilisés pour positionner l’objectif de recouvrement. (G) Les boutons haut et bas utilisés pour positionner l’objectif de détection. Notez que la télécommande n’est pas nécessaire, et toutes ces manipulations peuvent être effectuées dans le logiciel. Cependant, il est pratique de pouvoir contrôler les objectifs et la position de la scène tout en regardant dans l’environnement de la scène du microscope. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S3 : Module de fluorescence pour piège optique. La source de lumière blanche à fluorescence PhotoFluor 89North est couplée à l’arrière du microscope inversé. Il est activé et désactivé avec un interrupteur à bascule (flèche). Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S4 : Tourelle cubique filtrante à fluorescence. La tourelle (flèche) peut être tournée pour utiliser le cube filtrant nécessaire à l’imagerie dans les colorants DIC, rhodamine ou Alexa Fluor 488. Notez que les cubes de filtre peuvent être remplacés pour personnaliser la configuration pour l’utilisation de différents fluorophores. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S5 : Logiciel Nanotracker2. (A) Bouton d’alimentation laser et commande. B) Fenêtre de positionnement objectif. Des flèches directionnelles sont utilisées pour déplacer les objectifs de détection (en haut) et de piégeage (en bas). Les flèches doubles déplacent les objectifs à une vitesse plus élevée. Le bouton bleu et rouge en bas à gauche détache les objectifs et les ramène à leur position initiale. Ceci est nécessaire lors du prélèvement d’échantillons dans et hors de l’étape du microscope. Le troisième bouton à partir de la gauche avec les objectifs et l’icône du cadenas « couple » les objectifs de sorte que lorsqu’ils sont tous les deux mis au point et atteignent l’éclairage de Kohler, l’utilisateur peut déplacer les objectifs de recouvrement et de détection de haut en bas dans l’axe z. (C) Fenêtre de positionnement de l’échantillon utilisée pour déplacer l’étage du microscope sur les axes x et y. Les flèches doubles déplacent la scène à une vitesse plus élevée. Cette fenêtre est activée en cliquant sur l’icône flèche haut/bas et gauche/droite dans le menu supérieur. (D) Fenêtre de visualisation de la caméra. L’icône de clé peut être utilisée pour définir des conditions d’imagerie personnalisées. Cette fenêtre est activée en cliquant sur l’icône Caméra dans le menu supérieur. E) Fenêtre d’éclairage du microscope. Cette fenêtre est activée en cliquant sur l’icône Ampoule dans le menu supérieur. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S6 : fenêtre d’étalonnage. (A) Cette fenêtre est utilisée pour l’étalonnage des perles et est activée en cliquant sur l’icône Cal dans le menu supérieur. Pour calibrer une perle, un meilleur ajustement de la fréquence d’angle est réalisé dans les signaux x, y et z. (B) Pour chaque signal, choisissez le bouton de signal approprié en haut à gauche. (C) Cliquez sur exécuter et optimisez l’ajustement en cliquant et en faisant glisser dans la fenêtre verte (D). (E) Une fois satisfait de l’ajustement, cliquez sur Utiliser pour la sensibilité et la rigidité. Cela permettra d’enregistrer le déplacement en nanomètres et la force en piconewtons. (F) Ensuite, cliquez sur Accepter les valeurs en bas à gauche. Répétez l’opération pour les directions y et z. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S7 : Fenêtre d’acquisition de données. Cette fenêtre est utilisée pour acquérir des données de position et de force et permet à l’utilisateur de voir les mesures en temps réel. (A) Cette fenêtre est activée en cliquant sur l’icône x,t dans le menu supérieur. (B) L’utilisateur peut basculer entre la visualisation des signaux x et y. (C) Cliquez sur Démarrer pour commencer à visualiser les données. Cliquez sur Enregistrement automatique pour enregistrer les données. Cliquez sur Démarrer l’enregistrement pour commencer l’enregistrement et la sauvegarde des données. Veuillez cliquer ici pour télécharger ce fichier.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Une étude in vitro utilisant des pincettes optiques combinées à l’imagerie par fluorescence a été réalisée pour étudier la dynamique des ensembles de myosine interagissant avec les filaments d’actine. Les faisceaux d’actine-myosine-actine ont été assemblés à l’aide de myosine musculaire II, d’actine rhodamine au bas du faisceau et sur la surface de la lame, et de filaments d’actine biotinylés marqués 488 sur le dessus du paquet. La protéine d’actine du muscle de lapin a été polymérisée et stabilisée à l’aide de tampons généraux d’actine (GAB) et de tampons de polymérisation d’actine (APB). Les GAB et APB doivent être fraîchement préparés chaque jour en laboratoire à l’aide d’ATP, de tampon FC et de tampon TC. La myosine musculaire II a été utilisée pour former les sandwichs actine-myosine-actine. La phalloïdine a été utilisée pour la coloration fluorescente des filaments d’actine, ainsi que pour la stabilisation in vitro.
L’activité de la myosine peut être confirmée en effectuant un test filamentaire de glissement standard tel que publié précédemment46,47. La myosine II et ses sous-fragments peuvent se lier à la surface de la lamelle de couverture dans une variété d’orientations, et la présence du domaine de la queue peut ralentir le glissement du filament par rapport aux essais utilisant de la méromyosine lourde46,48,49. Cependant, le glissement et le mouvement de surface peuvent encore être observés. Une démonstration plus apparente de l’activité de la myosine est la rupture active du filament d’actine qui peut être observée lorsque des filaments d’actine plus longs sont brisés en fragments plus petits qui glissent ensuite dans plusieurs directions. Cela se produit en raison de la forte concentration de moteurs actifs à la surface, a été observée par plusieurs laboratoires et ne se produit pas sans moteurs actifs de myosine présents 42,50,51,52,53,54. De plus, le test du faisceau présenté ici aide à atténuer les problèmes de motilité qui ont été principalement associés au test du filament planant, tels que la variété des orientations de liaison du moteur sur un couvercle en verre, car le test du faisceau implique le blocage de la caséine de la surface du verre de sorte que les moteurs se lient dans le faisceau 47,55,56.
La première étape consiste à ajouter des filaments d’actine de rhodamine comme filament inférieur ou modèle à une lamelle de couverture revêtue de poly-L-lysine dans une cellule d’écoulement. La poly-L-lysine est utilisée pour favoriser la liaison à l’actine puisque la polylysine est chargée positivement tandis que l’actine a des charges négatives et a été utilisée dans des préparations de tests in vitro cytosquelettiques antérieures61,62,63. Avant la formation du faisceau, différentes dilutions d’actine ont été ajoutées à une cellule d’écoulement pour optimiser la concentration d’actine. Dans ce cas, 600x du stock était la dilution optimale qui a donné un nombre suffisant de filaments matriciels pour la formation de faisceaux, mais avec un espacement adéquat pour que les faisceaux soient individualisés. La dilution a été effectuée à l’aide du tampon APB. L’ajout d’actine rhodamine a été suivi d’une couche de caséine pour bloquer la surface et éviter une liaison non spécifique. La cellule d’écoulement a été incubée pendant 30 minutes et lavée après incubation avec un tampon pour éliminer tous les filaments d’actine non liés. Enfin, une combinaison de myosine, d’actine 488/biotine et de billes enrobées de streptavidine a été ajoutée à la cellule d’écoulement pour faciliter la formation de faisceaux actine-myosine. La concentration du cordon doit être telle qu’il y en ait suffisamment pour lier les faisceaux liés à la surface et suffisamment en suspension pour faciliter l’étalonnage. Cependant, une concentration trop élevée d’un cordon peut causer des difficultés lors des expériences de piégeage en raison des perles voisines tombant dans le piège laser et perturbant la mesure. Des moteurs de myosine sont ajoutés à la combinaison juste avant de l’injecter sur la lame afin que les moteurs de myosine ne s’agrègent pas de manière préventive avec la cargaison ou le filament d’actine biotinylé supérieur et lieront ainsi la rhodamine inférieure pour regrouper les filaments d’actine biotinylés.
Le système de piégeage optique NT2 est un piège optique commercial avec des modalités combinées d’imagerie par fond clair, contraste d’interférence différentielle (DIC) et épifluorescence. Il est couplé à un microscope inversé Zeiss AxioObserver 3 avec des objectifs de piégeage et de détection par immersion dans l’eau 100x/NA 1.46 et 63x/NA 1.0. Le système est équipé de la capacité de capture par clic et glissement d’un piège laser et peut être utilisé lors de l’imagerie dans l’une des modalités énumérées précédemment. Les faisceaux formés sont détectés et confirmés à l’aide de l’imagerie par fluorescence. Le fait d’avoir une source de lumière blanche avec des cubes filtrants appropriés (GFP/FITC et TRITC/CY3) permet une commutation rapide entre l’imagerie filamentaire. Les AF colocalisés ont été vérifiés en visualisant les AF aux différentes longueurs d’onde d’excitation avant de prendre chaque mesure de force à l’aide d’une pince optique. Comme les filaments peuvent photoblanchir rapidement, même avec un réactif de piégeage de l’oxygène, il est suggéré que les chercheurs optimisent les paramètres de visualisation tels que l’intensité et le temps d’exposition avant d’effectuer les expériences de faisceau.
Le piégeage optique a été utilisé pour prendre les mesures de force, en utilisant les billes de streptavidine en présence d’ATP pour lier le filament d’actine de cargaison biotinylé et activer la génération de force de myosine comme transducteur de force. Les données sur le déplacement et la force en fonction du temps obtenues par piégeage optique ont été extraites du logiciel de piégeage aux fins d’analyse. Cependant, le logiciel de piégeage commercial fournit également des routines d’analyse qui peuvent être utilisées, ou des algorithmes personnalisés dans d’autres programmes peuvent être programmés par l’utilisateur pour visualiser et analyser les données de piégeage. Sur les systèmes de piégeage optique personnalisés, l’utilisateur peut avoir des lasers d’excitation au lieu d’une source de lumière blanche avec des filtres, qui sont également acceptables à utiliser. De plus, les colorants fluorescents peuvent être modifiés pour être adaptés à l’équipement existant qu’un utilisateur peut avoir si les spectres d’émission ne se chevauchent pas et ne provoquent pas de saignement.
Nous notons que le test présenté est un test de base qui peut être personnalisé par l’utilisateur en fonction de sa question de recherche dans le domaine de la mécanique d’ensemble de l’actomyosine. Le flux de travail général peut également être appliqué à d’autres systèmes d’ensemble cytosquelettique in vitro qui peuvent être intéressants, tels que les tests de faisceaux de microtubules qui forment des modèles minimaux de fuseau mitotique 32,61,63,64,65,66. Les modifications pourraient inclure, sans toutefois s’y limiter, la modification des étiquettes fluorophores adaptées à la configuration existante de l’utilisateur; modification de la concentration, de la construction ou de l’isotype de myosine; et le titrage des conditions tampons, entre autres aspects.
Des défis potentiels sont possibles lors de la réalisation de ce test. Lors de la formation des faisceaux actine-myosine, la concentration de myosine dans les faisceaux d’actine peut ne pas être homogène sur toute la lame. Pour tenir compte de cela, plusieurs faisceaux sur l’ensemble de la diapositive seront mesurés pour s’assurer que les profils de distribution motrice et de génération de force sont correctement échantillonnés. Il est également difficile de connaître l’orientation des paquets si cela est nécessaire pour interpréter les données sur les forces. Par conséquent, plusieurs essais doivent être effectués pour chaque faisceau. On pourrait également incorporer l’étiquetage de l’extrémité du filament d’actine à l’aide de gelsoline fluorescente ou de billes enrobées de gelsoline d’une taille plus petite que la poignée de piégeage optique. L’imagerie par fluorescence peut également être utilisée pour examiner les forces des composantes x et y afin de déduire l’orientation du faisceau. De plus, comme l’état d’agrégation de la myosine est fortement influencé par la force ionique du tampon avec formation de filaments épais se produisant lors d’une dilution rapide de KCl, la concentration de sel tampon doit être surveillée de manière appropriée67,68.
Des études antérieures qui utilisaient d’autres méthodes in vitro telles que les tests de glissement ont été utiles pour identifier le rôle des domaines de myosine et étudier la configuration et les interactions entre la myosine et d’autres protéines de liaison à l’actine. Cependant, ces méthodes présentent un inconvénient en ce sens que la liaison de la myosine sur une surface rigide limitera le potentiel de coordination entre les moteurs de myosine et donc la rétroaction de mécanodétection qui se produit pour déterminer si l’ensemble moteur est dans un mode de rapport de service élevé ou faible 33,35,41,69. De plus, le piégeage optique avec des réseaux moteurs à myosine unique ne permet pas de comprendre clairement comment les moteurs de myosine interagissent entre eux et avec les filaments d’actine. Le protocole développé ici permet d’étudier la dynamique de l’ensemble moteur de la myosine au sein d’un réseau d’actine hiérarchique conforme. Il est également personnalisable en termes de caractéristiques de l’ensemble moteur-filament telles que la concentration, l’isoforme et l’environnement tampon, entre autres aspects, pour permettre une étude systématique. Le protocole présenté est une plate-forme pour les études futures de réseaux d’actomyosine plus complexes et maintient la précision des mesures de déplacement et de génération de force facilitée par le piégeage optique traditionnellement utilisé pour les études sur une seule molécule.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs n’ont aucun conflit d’intérêts à déclarer.
Acknowledgments
Ce travail est soutenu en partie par le Graduate Student Council Research Fellowship (OA) de l’Université du Mississippi, le Sally McDonnell-Barksdale Honors College de l’Université du Mississippi (JCW, JER), le Mississippi Space Grant Consortium sous le numéro de subvention NNX15AH78H (JCW, DNR) et l’American Heart Association sous le numéro de subvention 848586 (DNR).
Materials
| Name | Company | Catalog Number | Comments |
| Actin protein (biotin): skeletal muscle | Cytoskeleton | AB07-A | Biotinylated actin protein |
| Actin protein, rabbit skeletal muscle | Cytoskeleton | AKL99-A | Actin protein |
| Alexa Fluor 488 Phalloidin | Invitrogen | A12379 | Actin stabilizer and Alexa Fluor 488 stain |
| ATP | Fisher scientific | BP413-25 | Required for actin assembly and myosin motility |
| Beta-D-glucose | Fisher scientific | MP218069110 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Blotting Grade Blocker (casein) | Biorad | 1706404 | Used to block surface from non-specific binding |
| CaCl2 | Fisher scientific | C79500 | Calcium chloride, provides the necessary control over the dynamics of actin myosin network |
| Catalase | Fisher scientific | ICN10040280 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Coverslips | Fisher scientific | 12544C | Used to make flow cells |
| DTT | Fisher scientific | AC327190010 | Used for buffer preparation |
| Ethanol | Fisher scientific | A4094 | Regent used for cleaning coverslips |
| Glucose oxidase | Fisher scientific | 34-538-610KU | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| KCl | Fisher scientific | P217-500 | Used for buffer preparation |
| KOH | Fisher scientific | P250-1 | Used to etch coverslips and adjust buffer pH |
| MgCl2 | Fisher scientific | M33-500 | Used for buffer preparation |
| Microscope slides | Fisher scientific | 12-544-2 | Used to make flow cells |
| Myosin II protein: rabbit skeletal muscle | Cytoskeleton | MY02 | Full length myosin motor protein isolated from rabbit skeletal muscle |
| Nanotracker2 | Bruker/JPK | NT2 | Optical trapping instrument |
| Poly-l-lysine | Sigma-Aldrich | P8920 | Facilities adhesion of actin filaments onto glass surface of the coverslip |
| Rhodamine Phalloidin | Cytoskeleton | PHDR1 | Actin stabilizer and rhodamine fluorescent stain |
| Streptavidin beads, 1 μm | Spherotech | SVP-10-5 | Optical trapping handle |
| Tris-HCl | Fisher scientific | PR H5121 | Used for buffer preparation |
References
- Goldstein, L. S. Kinesin molecular motors: transport pathways, receptors, and human disease. Proceedings of the National Academy of Sciences of the United States of America. 98 (13), 6999-7003 (2001).
- Lee Sweeney, H., Holzbaur, E. L. F.
- O'Connell, C. B., Tyska, M. J., Mooseker, M. S. Myosin at work: Motor adaptations for a variety of cellular functions. Biochimica et Biophysica Acta - Molecular Cell Research. 1773 (5), 615-630 (2007).
- Kaya, M., Tani, Y., Washio, T., Hisada, T., Higuchi, H. Coordinated force generation of skeletal myosins in myofilaments through motor coupling. Nature Communications. 8, 1-13 (2017).
- Akhshi, T. K., Wernike, D., Piekny, A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton. 71 (1), 1-23 (2014).
- Brawley, C. M., Rock, R. S. Unconventional myosin traffic in cells reveals a selective actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America. 106 (24), 9685-9690 (2009).
- Hartman, M. A., Spudich, J. A. The myosin superfamily at a glance. Journal of Cell Science. 125 (7), 1627-1632 (2012).
- Spudich, J. A., et al.
- Sommese, R. F., et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proceedings of the National Academy of Sciences of the United States of America. 110 (31), 12607-12612 (2013).
- Nag, S., et al. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nature Structural & Molecular Biology. 24 (6), 525-533 (2017).
- Kawana, M., Sarkar, S. S., Sutton, S., Ruppel, K. M., Spudich, J. A. Biophysical properties of human b-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Science Advances. 3 (2), 1-11 (2017).
- Girolami, F., et al. Novel α-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: A massively parallel sequencing study. Circulation: Cardiovascular Genetics. 7 (6), 741-750 (2014).
- Debold, E. P., et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. American Journal of Physiology - Heart and Circulatory Physiology. 293 (1), 284-291 (2007).
- Barron, J. T.
- Duke, T. A. J.
- Vilfan, A., Duke, T.
- Huxley, A. F. Muscle structure and theories of contraction. Progress in Biophysics and Biophysical Chemistry. 7, 255-318 (1957).
- Huxley, H. E. Fifty years of muscle and the sliding filament hypothesis. European Journal of Biochemistry. 271 (8), 1403-1415 (2004).
- Kad, N. M., Kim, S., Warshaw, D. M., VanBuren, P., Baker, J. E. Single-myosin crossbridge interactions with actin filaments regulated by troponin-tropomyosin. Proceedings of the National Academy of Sciences of the United States of America. 102 (47), 16990-16995 (2005).
- Veigel, C., Molloy, J. E., Schmitz, S., Kendrick-Jones, J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nature Cell Biology. 5 (11), 980-986 (2003).
- Spudich, J. A.
- Simmons, R. M., Finer, J. T., Chu, S., Spudich, J. A. Quantitative measurements of force and displacement using an optical trap. Biophysical Journal. 70 (4), 1813-1822 (1996).
- Finer, J. T., Simmons, R. M., Spudich, J. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 368 (6467), 113-119 (1994).
- Kron, S. J., Uyeda, T. Q. P., Warrick, H. M., Spudich, J. A. An approach to reconstituting motility of single myosin molecules. Journal of Cell Science. 98, 129-133 (1991).
- Molloy, J. E., Burns, J. E., Kendrick-Jones, B., Tregear, R. T., White, D. C. S. Movement and force produced by a single myosin head. Nature. 378 (6553), 209-212 (1995).
- Ruegg, C., et al. Molecular motors: Force and movement generated by single Myosin II molecules. Physiology. 17 (5), 213-218 (2002).
- Nayak, A., et al. Single-molecule analysis reveals that regulatory light chains fine-tune skeletal myosin II function. Journal of Biological Chemistry. 295 (20), 7046-7059 (2020).
- Dupuis, D. E., Guilford, W. H., Wu, J., Warshaw, D. M.
- Tyska, M. J., et al. Two heads of myosin are better than one for generating force and motion. Proceedings of the National Academy of Sciences of the United States of America. 96 (8), 4402-4407 (1999).
- Tyska, M. J., Warshaw, D. M.
- Finer, J. T., et al.
- Al Azzam, O., Trussell, C. L., Reinemann, D. N. Measuring force generation within reconstituted microtubule bundle assemblies using optical tweezers. Cytoskeleton. 78 (3), 111-125 (2021).
- Wagoner, J. A., Dill, K. A. Evolution of mechanical cooperativity among myosin II motors. Proceedings of the National Academy of Sciences of the United States of America. 118 (20), 2101871118 (2021).
- Walcott, S., Warshaw, D. M., Debold, E. P. Mechanical coupling between myosin molecules causes differences between ensemble and single-molecule measurements. Biophysical Journal. 103 (3), 501-510 (2012).
- Stewart, T. J., Murthy, V., Dugan, S. P., Baker, J. E. Velocity of myosin-based actin sliding depends on attachment and detachment kinetics and reaches a maximum when myosin-binding sites on actin saturate. Journal of Biological Chemistry. 297 (5), 101178 (2021).
- Hilbert, L., Cumarasamy, S., Zitouni, N. B., Mackey, M. C., Lauzon, A. M. The kinetics of mechanically coupled myosins exhibit group size-dependent regimes. Biophysical Journal. 105 (6), 1466-1474 (2013).
- Debold, E. P., Walcott, S., Woodward, M., Turner, M. A. Direct observation of phosphate inhibiting the Force-generating capacity of a miniensemble of myosin molecules. Biophysical Journal. 105 (10), 2374-2384 (2013).
- Kaya, M., Higuchi, H. Nonlinear elasticity and an 8-nm working stroke of single myosin molecules in myofilaments. Science. 329 (5992), 686-689 (2010).
- Pertici, I., et al. A myosin II nanomachine mimicking the striated muscle. Nature Communications. 9 (1), 1-10 (2018).
- Cheng, Y. S., De Souza Leite, F., Rassier, D. E. The load dependence and the force-velocity relation in intact myosin filaments from skeletal and smooth muscles. American Journal of Physiology - Cell Physiology. 318 (1), 103-110 (2020).
- Stam, S., Alberts, J., Gardel, M. L., Munro, E. Isoforms confer characteristic force generation and mechanosensation by myosin II filaments. Biophysical Journal. 108 (8), 1997-2006 (2015).
- Rastogi, K., Puliyakodan, M. S., Pandey, V., Nath, S., Elangovan, R. Maximum limit to the number of myosin II motors participating in processive sliding of actin. Scientific Reports. 6, 1-11 (2016).
- Debold, E. P., Patlak, J. B., Warshaw, D. M. Slip sliding away: Load-dependence of velocity generated by skeletal muscle myosin molecules in the laser trap. Biophysical Journal. 89 (5), 34-36 (2005).
- Albert, P. J., Erdmann, T., Schwarz, U. S. Stochastic dynamics and mechanosensitivity of myosin II minifilaments. New Journal of Physics. 16, (2014).
- Erdmann, T., Schwarz, U. S. Stochastic force generation by small ensembles of myosin II motors. Physical Review Letters. 108 (18), 1-5 (2012).
- Guo, B., Guilford, W. H. The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motility and the Cytoskeleton. 59 (4), 264-272 (2004).
- Miller-Jaster, K. N., Petrie Aronin, C. E., Guilford, W. H. A quantitative comparison of blocking agents in the in vitro motility assay. Cellular and Molecular Bioengineering. 5 (1), 44-51 (2012).
- Mansoon, A., Balaz, M., Albet-Torres, N., Rosengren, K. J. In vitro assays of molecular motors -- impact of motor-surface interactions. Frontiers in Bioscience. 13, 5732-5754 (2008).
- Persson, M., et al. Heavy meromyosin molecules extending more than 50 nm above adsorbing electronegative surfaces. Langmuir. 26 (12), 9927-9936 (2010).
- Kron, S. J., Spudich, J. A. Fluorescent actin filaments move on myosin fixed to a glass surface. Proceedings of the National Academy of Sciences of the United States of America. 83 (17), 6272-6276 (1986).
- Yanagida, T., Nakase, M., Nishiyama, K., Oosawa, F. Direct observation of motion of single F-actin filaments in the presence of myosin. Nature. 307 (5946), 58-60 (1984).
- Tsuda, Y., Yasutake, H., Ishijima, A., Yanagida, T. Torsional rigidity of single actin filaments and actin-actin bond breaking force under torsion measured directly by in vitro micromanipulation. Proceedings of the National Academy of Sciences of the United States of America. 93 (23), 12937-12942 (1996).
- Stewart, T. J., et al. Actin sliding velocities are influenced by the driving forces of actin-myosin binding. Cellular and Molecular Bioengineering. 6 (1), 26-37 (2013).
- Harada, Y., Sakurada, K., Aoki, T., Thomas, D. D., Yanagida, T. Mechanochemical coupling in actomyosin energy transduction by in vitro movement assay. Journal of Molecular Biology. 216 (1), 49-68 (1990).
- Fordyce, P. M., Valentine, M. T., Block, S. M. Advances in surface-based assays for single molecules. Single-Molecule Techniques: A Laboratory Manual. , 431-460 (2008).
- Ozeki, T., et al. Surface-bound casein modulates the adsorption and activity of kinesin on SiO2 surfaces. Biophysical Journal. 96 (8), 3305-3318 (2009).
- Neuman, K. C., Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nature Methods. 5 (6), 491-505 (2008).
- Neuman, K. C., Block, S. M.
- Thoresen, T., Lenz, M., Gardel, M. L. Thick filament length and isoform composition determine self-organized contractile units in actomyosin bundles. Biophysical Journal. 104 (3), 655-665 (2013).
- Matusovsky, O. S., et al. Millisecond conformational dynamics of skeletal Myosin II power stroke studied by high-speed atomic force microscopy. ACS Nano. 15 (2), 2229-2239 (2021).
- Reinemann, D. N., et al. Collective force regulation in anti-parallel microtubule gliding by dimeric Kif15 kinesin motors. Current Biology. 27 (18), 2810-2820 (2017).
- Cordova, J. C., et al. Bioconjugated core-shell microparticles for high-force optical trapping. Particle and Particle Systems Characterization. 35 (3), 1-8 (2018).
- Reinemann, D. N., Norris, S. R., Ohi, R., Lang, M. J. Processive Kinesin-14 HSET exhibits directional flexibility depending on motor traffic. Current Biology. 28 (14), 2356-2362 (2018).
- Forth, S., Hsia, K. C., Shimamoto, Y., Kapoor, T. M. Asymmetric friction of nonmotor MAPs can lead to their directional motion in active microtubule networks. Cell. 157 (2), 420-432 (2014).
- Shimamoto, Y., Kapoor, T. M. Analyzing the micromechanics of the cell division apparatus. Methods in Cell Biology. 145, 173-190 (2018).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Thoresen, T., Lenz, M., Gardel, M. L.
- Murrell, M., Thoresen, T., Gardel, M.
- Weirich, K. L., Stam, S., Munro, E., Gardel, M. L. Actin bundle architecture and mechanics regulate myosin II force generation. Biophysical Journal. 120 (10), 1957-1970 (2021).
