

Summary
An ex vivo protocol to generate mature human red blood cells from hematopoietic stem/progenitors is described. Additionally we describe an efficient lentiviral-delivery method to knockdown the transcription factor TAL1 in primary erythroid cells. The efficiency of lentivirus mediated gene delivery is demonstrated using GFP expressing viruses.
Abstract
Erythropoiesis is a commonly used model system to study cell differentiation. During erythropoiesis, pluripotent adult human hematopoietic stem cells (HSCs) differentiate into oligopotent progenitors, committed precursors and mature red blood cells 1. This process is regulated for a large part at the level of gene expression, whereby specific transcription factors activate lineage-specific genes while concomitantly repressing genes that are specific to other cell types 2. Studies on transcription factors regulating erythropoiesis are often performed using human and murine cell lines that represent, to some extent, erythroid cells at given stages of differentiation 3-5. However transformed cell lines can only partially mimic erythroid cells and most importantly they do not allow one to comprehensibly study the dynamic changes that occur as cells progress through many stages towards their final erythroid fate. Therefore, a current challenge remains the development of a protocol to obtain relatively homogenous populations of primary HSCs and erythroid cells at various stages of differentiation in quantities that are sufficient to perform genomics and proteomics experiments.
Here we describe an ex vivo cell culture protocol to induce erythroid differentiation from human hematopoietic stem/progenitor cells that have been isolated from either cord blood, bone marrow, or adult peripheral blood mobilized with G-CSF (leukapheresis). This culture system, initially developed by the Douay laboratory 6, uses cytokines and co-culture on mesenchymal cells to mimic the bone marrow microenvironment. Using this ex vivo differentiation protocol, we observe a strong amplification of erythroid progenitors, an induction of differentiation exclusively towards the erythroid lineage and a complete maturation to the stage of enucleated red blood cells. Thus, this system provides an opportunity to study the molecular mechanism of transcriptional regulation as hematopoietic stem cells progress along the erythroid lineage.
Studying erythropoiesis at the transcriptional level also requires the ability to over-express or knockdown specific factors in primary erythroid cells. For this purpose, we use a lentivirus-mediated gene delivery system that allows for the efficient infection of both dividing and non-dividing cells 7. Here we show that we are able to efficiently knockdown the transcription factor TAL1 in primary human erythroid cells. In addition, GFP expression demonstrates an efficiency of lentiviral infection close to 90%. Thus, our protocol provides a highly useful system for characterization of the regulatory network of transcription factors that control erythropoiesis.
Protocol
Part I. ex vivo erythropoiesis of human hematopoietic stem/progenitor cells
1. Isolation of CD34+ human hematopoietic stem/progenitor cells
Human CD34+ cell population, which contains a mixture of hematopoietic stem cells (HSCs) and early progenitors 8, is harvested from umbilical cord blood, peripheral blood mobilized with G-CSF (leukapheresis) or bone marrow (Figure 1-Step1). If using cord blood or peripheral blood, go directly to step 1.2. If using bone marrow, start at step 1.1. For reagents and equipment, see Table 1.
This step is done under sterile conditions (i.e. hood) and at RT unless indicated otherwise.
- Prepare a single cell suspension of bone marrow:
- Resuspend the marrow in 10 vol. of RPMI 1640 containing 0.02% collagenase B and 100 U/ml DNase 1
- Mix gently on a balance shaker for 45 min.
- Filter cells through a 30μm nylon mesh.
- Isolate light-density mononuclear cells (MNCs) by Ficoll density gradient centrifugation:
- Dilute blood or marrow suspension with 2-4 vol. of PBS containing 2% fetal bovine serum (FBS) and 2mM EDTA. N.B. Higher volumes increase the purity of MNCs.
- In a 50 ml conical tube carefully layer 35 ml of diluted cell suspension over 15 ml of Ficoll-paque. Centrifuge at 400g for 30 min. in a swinging-bucket rotor with no brake.
- Aspirate the upper layer leaving the intermediate MNC layer undisturbed.
- Transfer the interphase layer (containing MNCs) to a new 50 ml conical tube.
- Add PBS containing 2% FBS and 2 mM EDTA to fill this tube, mix gently by inverting the tube and centrifuge at 300g for 10 min. at RT. Discard supernatant.
- Wash the cell pellet 2 additional times to remove platelets. N.B. This step increases the purity of CD34+ cells.
- Take 20 μl of cell suspension and place in an eppendorf tube. Add 200 μl of Red Blood Cells Lysis Buffer (Table 1) and incubate at RT for 2 min. Place 18 μl of this suspension under a hemocytometer and count the nucleated MNCs.
- MNCs can be used immediately or frozen at 108 cell/ml in cell culture medium containing 10% DMSO and stored in liquid nitrogen.
- Isolate CD34+ cells by positive immunomagnetic selection (Figure 1-Step 1)
We use the CD34 Microbead Kit (containing a solution of Microbeads conjugated to monoclonal mouse human CD34 antibodies and a solution of FcR Blocking Reagent containing human IgG) in combination with two LS Mini-Macs columns, the autoMACS Running Buffer and a MiniMACS Separator (Table 1). Below we describe our protocol starting from 2.3x108 MNCs. If different numbers of cells are used, volumes should be scaled-up or down accordingly. In addition, for up to 2x108 MNCs a MS MiniMacs column can be used instead of the LS MiniMacs column.
Keep cells and reagents cold (4-8°C) to prevent capping of antibodies on the cell surface and non-specific cell labeling.- Resuspend the pellet containing 2.3x108 MNCs in 690 μl of Running Buffer (300 μl per 108 MNCs where 300 μl is the minimum suspension volume required) and transfer into a 15 ml conical tube.
- Add 230 μl of FcR Blocking Reagent
- Add 230 μl of CD34 MicroBeads solution.
- Mix well by hand and incubate for 30 min. at 4°C.
- Wash the cells by adding 10 ml Running Buffer and centrifuge at 300g for 10 min.
- Aspirate the supernatant completely.
- Resuspend the cells in 1.2 ml Running Buffer (use 0.5 ml buffer per 108 MNCs) N.B. It is important to obtain a single-cell suspension before magnetic separation. If needed, pass cells through a 30μm nylon mesh to remove cell clumps.
- Prepare the LS column by placing it in the magnetic field of a MiniMACS Separator.
- Rinse the column with 3 ml Running Buffer.
- Apply 1.2 ml of cell suspension onto the column.
- Collect the flow-through and use it to verify the yield and purity of the CD34+ cell isolation. Alternatively freeze the cells contained in this fraction and store them in liquid nitrogen. N.B This fraction contains unlabeled cells and can also contain some labeled CD34+ cells that did not attach to the column.
- Wash the column three times with 3 ml of Running Buffer, combine the washes, and use this fraction similarly to the flow-through fraction above.
- Remove the column from the magnet and place it on a 15 ml polypropylene collection tube.
- To elute cells, add 5 ml of Running Buffer onto the column and immediately flush out the labeled CD34+ cells by firmly pushing the plunger into the column.
- Repeat the column purification procedure (steps h to n) by passing the cells over a freshly prepared LS column. N.B. This increases the purity of CD34+ cells. From a suspension of 2.3x108 MNCs isolated from leukapheresis, we typically obtain 1.1x106 CD34+ cells at a purity > 95±3% (Figure 1-Step 1).
- Freeze the cells in culture medium containing 10% DMSO and store in liquid nitrogen or proceed directly to step 2).
2. Erythroid differentiation in cell culture
CD34+ cells are cultured in supplemented IMDM medium with sequential supply of hydrocortisone and specific combinations of cytokines according to a four-step protocol adapted from 6 (Figure 1-Step 2). A series of tests is performed throughout the procedure to verify cell growth and erythroid differentiation.
The number of CD34+ cells required to start a culture is variable, depending on the scale of the experiment. Typically, for
gene expression studies we perform a small-scale culture starting with 0.1x106 CD34+ cells in 10 ml medium and maintaining small volumes by freezing extra
cells throughout the procedure. For studies where protein extraction is required, cultures are performed at a larger scale. Here we describe a large-scale
experiment where we start with ~7x106 CD34+ cells isolated from leukapheresis.
Sterile conditions are used throughout. See Tables 1, 2 and 3 for reagents.
- Prepare reagents
- Prepare the supplemented IMDM medium as described in Table 2 and freeze aliquots of 40 ml at -20°C (can be done in advance).
- Prepare stock solutions of hydrocortisone and cytokines as described in Table 3.
- Step 1: Day 0-11
Hematopoietic cells are grown in supplemented IMDM medium containing hydrocortisone (HC), stem cell factor (SCF), interleukin 3 (IL-3) and erythropoietin (EPO) (Table 3). MS-5 murine bone marrow stromal cells (Table 1) are amplified in preparation for Step 2.
- Day 0:
- Pre-warm 120 ml of supplemented IMDM medium and add HC, SCF, IL-3 and EPO (Table 3).
- Thaw 7x106 CD34+ cells by first placing the vial(s) in a 37°C water bath. While the cells are still frozen, add 1 ml of pre-warmed medium and transfer the cells to a 15 ml conical tube. Wash the vial with 2 ml medium and transfer to the 15 ml tube.
- Add 6 ml medium to the cell suspension, mix by inverting the tube and spin at 200 g for 5 min.
- Aspirate the supernatant and resuspend the cell pellet in 10 ml fresh medium.
- Count living cells using a hemocytometer after mixing 10 μl of cell suspension with 10 μl Trypan Blue solution (Table 1). NB: Cell death is about 25% during thawing, so the concentration of live cells should be ~0.5x106 cell/ml.
- Save approximately 3 ml of cell suspension for tests (see paragraph 6).
- Transfer the remaining 7 ml into a T-175 flask and add 63 ml of medium to have 0.05x106 cell/ml in 70 ml and place the flask in an incubator at 37°C.
- Day 1:
- Pre-warm 85 ml of MS-5 cell culture medium: αMEM with Glutamax (Table 1), 10% FBS and 100 U/ml of penicillin and 100 μg/ml of streptomycin.
- Prepare three 150mm x 25mm round culture dishes, each seeded with 2x106 MS-5 cell in 25 ml medium to obtain 0.08x106 cell/ml and place in a 37°C incubator. NB: Best results are obtained using low passage (<20) MS-5 cells. Confluent culture should be expanded by splitting 1:5 every third day using trypsin containing 0.05% EDTA.
- Day 2:
- Count hematopoietic cells with Trypan blue to exclude dead cells, and harvest the necessary cells (i.e. ~20 ml of culture) for tests (see paragraph 6). NB: In our experience, during the first 2 days, cell death might exceed 30-50% and as such the final concentration of the cells is usually close to that at Day 0 (Figure 2).
- Replace MS-5 culture medium with 25 ml of fresh medium making sure MS-5 cells that did not attach are removed.
- Day 4:
- Count hematopoietic cells with Trypan blue to exclude dead cells, and harvest the necessary cells (i.e. ~10 ml of culture) for tests (see paragraph 6). NB: At this point, cells should have started to proliferate with >75% of live cells at a concentration of ~0.2x106 cell/ml. Cells may be frozen at 1-2x106 cell/ml in supplemented IMDM medium containing 10% DMSO and stored in liquid nitrogen.
- Transfer the remaining ~40 ml of culture in a T-175 flask and add enough supplemented IMDM medium containing HC, SCF, IL-3 and EPO (Table 3) to reach a final volume of 150 ml. NB: Cell concentration should be ~0.05x106 cell/ml.
- Expand MS-5 cells (that should be 60-70% confluent) by diluting them 1:5 into new 150mm x 25 mm round culture dishes using trypsin 0.05% EDTA. Final cell concentration should be ~0.08x106 cell/ml. NB: Usually, one dish can be expanded 5 times such that you should have 15 dishes of MS-5 cells at ~0.08x106 cell/ml.
- Day 6:
- Count hematopoietic cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~5 ml of culture) for tests (see paragraph 6). NB: Cells are in a highly proliferating stage and the cell concentration should be maintained ≤1.5x106 cell/ml. If the cell concentration is higher, add fresh supplemented IMDM medium containing HC, SCF, IL-3 and EPO.
- Day 7:
- Count hematopoietic cells with Trypan blue to exclude dead cells. NB: In our experience, cells usually reach a density of 1.5x106 cell/ml at Day7.
- Add enough medium to maintain a cell concentration of ~0.8x106 cell/ml. NB: We usually add 100 ml of fresh supplemented IMDM medium containing HC, SCF, IL-3 and EPO to obtain 245 ml of cells at ~0.8x106 cell/ml.
- Expand MS-5 cells (that should be 60-70% confluent) by diluting them 1:5 into T-175 flasks (instead of dishes) using trypsin 0.05% EDTA. Final cell concentration should be ~0.08x106 cell/ml. NB: 61 flasks of MS-5 cells at ~0.08x106 cell/ml should be prepared. Freeze the remaining MS-5 cells and store them in liquid nitrogen.
- Day 8:
- Count hematopoietic cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~5 ml of culture) for tests (see paragraph 6). NB: Cell density should be ~1x106 cell/ml.
- Pre-warm 2L of supplemented IMDM medium containing SCF and EPO only (Tables 2 and 3).
- Harvest 200 ml of hematopoietic culture (containing ~200x106 cells), spin at 200 g for 10 min., aspirate the supernatant and resuspend the cell pellet in 40 ml of supplemented IMDM medium containing SCF and EPO.
- Distribute the cell suspension into 8 T-175 flasks (5 ml per flask) and add 245 ml of supplemented IMDM medium containing SCF and EPO to each flask. NB: We now have 8 flasks, each containing 250 ml of cells at 0.1x106 cell/ml.
- Remaining cells (~40ml) at Day 8 may be frozen at 1-2x106 cell/ml in supplemented IMDM medium containing 10% DMSO and stored in liquid nitrogen.
- Day 10:
- Count hematopoietic cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~5 ml of culture) for tests (see paragraph 6). NB: Cell density should be ~1x106 cell/ml.
- Day 0:
- Step 2: Day 11-15
Approximately half the cells are harvested for future use (e.g. nuclear extraction). The remaining erythroid cells are co-cultured on MS-5 cells in supplemented IMDM medium containing EPO (Tables 2 and 3). Erythroid cells must be transferred to a fresh MS-5 layer every 3-4 days.
- Day 11:
- Count erythroid cells with Trypan blue to exclude dead cells. NB: Cell density should be ~1.5x106 cell/ml.
- Pre-warm 1L of 1xPBS, 400ml of supplemented IMDM medium and 5L of supplemented IMDM medium containing EPO (Tables 2 and 3) at 37°C.
- Reserve a volume of culture equivalent to 15x108 cells (e.g. ~4 flasks of 250 ml each) and proceed to e.
- Remaining cells (e.g. 15x108) are harvested in conical tubes, centrifuged at 450 g for 10 min., resuspended in cold MS30 buffer (Table 5) containing 0.1mM DTT using a maximum of 108 cell/ml and stored at -80°C after snap-freeze in liquid nitrogen for future use (e.g. nuclear extract).
- Wash 50 flasks of confluent MS-5 cells with 20 ml of 1xPBS each and immediately add 80 ml of fresh supplemented IMDM medium containing EPO in each flask. NB: Because of the large number of flasks, we process 4 flasks at a time. Precisely, we retrieve 4 flasks of confluent MS-5 cells from the incubator, wash them with 1xPBS, add the 80 ml medium and return the flasks to the incubator. When all the flasks are prepared and returned to the incubator, we proceed to step f. Because of the number of flasks to be processed for a large-scale procedure, it is beneficial to have these steps performed by two persons working simultaneously.
- Centrifuge the equivalent of 15x108 erythroid cells in 4 conical tubes at 450 g for 10 min. and discard the supernatant.
- Wash the 4 cell pellets with 100 ml each of supplemented IMDM medium.
- Resuspend each cell pellet in 250 ml of supplemented IMDM medium containing EPO and distribute 20 ml of this cell suspension to each of the 50 flasks containing MS-5 cells and 80ml of medium. NB: At this step, you should have 50 flasks with confluent MS-5 cells containing 100 ml of erythroid cells at 0.3x106 cell/ml.
- Expand the remaining 11 flasks of MS-5 cells by diluting them 1:5 into T-175 flasks using trypsin 0.05% EDTA. Final cell concentration should be ~0.08x106 cell/ml. NB: 55 flasks of MS-5 cells at ~0.08x106 cell/ml should be prepared.
- Day 12:
- Count erythroid cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~4 ml of culture) for tests (see paragraph 6). NB: Cell density should be ~0.6x106 cell/ml.
- Day 14:
- Count erythroid cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~3 ml of culture) for tests (see paragraph 6). NB: Cell density should be ~0.9x106 cell/ml.
- Day 11:
- Step 3: Day 15-18
Some cells are harvested for future use (e.g. nuclear extraction). The remaining erythroid cells are co-cultured on MS-5 cells in supplemented IMDM medium with no cytokines.- Day 15:
- Count erythroid cells with Trypan blue to exclude dead cells. NB: Cell density should be ~1x106 cell/ml.
- Pre-warm 1L of 1xPBS and 5L of supplemented IMDM medium (Table 2) at 37°C.
- Reserve a volume of culture equivalent to 10x108 cells (e.g. ~10 flasks of 100 ml each) and proceed to e.
- Remaining cells (e.g. 40x108) are harvested and resuspended in cold MS30 buffer (Table 5) containing 0.1mM DTT for future use (see Step 2: Day 11 point d).
- Wash 50 flasks of confluent MS-5 cells with 20 ml of 1xPBS each and immediately add 80 ml of fresh supplemented IMDM medium in each flask.
- Centrifuge the equivalent of 10x108 erythroid cells in 4 conical tubes at 450 g for 10 min. and discard the supernatant.
- Wash the 4 cell pellets with 100 ml each of supplemented IMDM medium.
- Resuspend each cell pellet in 250 ml of supplemented IMDM medium and distribute 20 ml of this cell suspension to each of the 50 flasks containing MS-5 cells and 80ml of medium. NB: At this step, you should have 50 flasks with confluent MS-5 cells containing 100 ml of erythroid cells at 0.2x106 cell/ml.
- Expand the remaining 5 flasks of MS-5 cells by diluting them 1:5 into T-175 flasks using trypsin 0.05% EDTA. Final cell concentration should be ~0.08x106 cell/ml. NB: 20 flasks of MS-5 cells at ~0.08x106 cell/ml should be prepared. Freeze the remaining MS-5 cells and store in liquid nitrogen.
- Day 16:
- Count erythroid cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~4 ml of culture) for tests (see paragraph 6). NB: Cell density should be ~0.6x106 cell/ml.
- Day 15:
- Step 4: Day 18-26
Erythroid cells are concentrated 3-4 fold to obtain 5-7x106 cell/ml final and maintained on MS-5 layer to allow maturation. 10% FBS is added in supplemented IMDM medium to allow a better preservation of cultured red blood cells (cRBCs).- Day 18:
- Count erythroid cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~2 ml of culture) for tests (see paragraph 6). NB: Cell density should be ~1.7x106 cell/ml.
- Pre-warm 300ml of 1xPBS and 1.5L of supplemented IMDM medium (Table 2) containing 10% FBS at 37°C.
- Wash 15 flasks of confluent MS-5 cells with 20 ml of 1xPBS each and immediately add 80 ml of fresh supplemented IMDM medium in each flask.
- Centrifuge erythroid cells (i.e. the equivalent of 85x108 cells) at 450 g for 10 min., combine and distribute the pellet into 4 conical tubes.
- Resuspend each cell pellet in 75 ml of supplemented IMDM medium containing 10% FBS and distribute 20 ml of this cell suspension to each of the 15 flasks containing MS-5 cells and 80ml of medium. NB: At this step, you should have 15 flasks with confluent MS-5 cells containing 100 ml of erythroid cells at 6x106 cell/ml.
- Expand the remaining 5 flasks of MS-5 cells by diluting them 1:5 into T-175 flasks using trypsin 0.05% EDTA. Final cell concentration should be ~0.08x106 cell/ml. NB: 20 flasks of MS-5 cells at ~0.08x106 cell/ml should be prepared. Freeze the remaining MS-5 cells and store in liquid nitrogen.
-
Day 20:
- Count erythroid cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~1.5 ml of culture) for tests (see paragraph 6). NB: Cell density is maintained ~1.5x106 cell/ml.
-
Day 22:
- Count erythroid cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~1.5 ml of culture) for tests (see paragraph 6). NB: Cell density is maintained ~1.5x106 cell/ml.
- Change medium and transfer erythroid cells on new layers of MS-5 cells as described for Day 18.
-
Day 24:
- Count erythroid cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~1.5 ml of culture) for tests (see paragraph 6). NB: Cell density is maintained ~1.5x106 cell/ml.
-
Day 26:
- Count erythroid cells with Trypan blue to exclude dead cells, and harvest the necessary number of cells (i.e. ~1.5 ml of culture) for tests (see paragraph 6). NB: Cell density is maintained ~1.5x106 cell/ml.
- Harvest non-adherent enucleated and fully hemoglobinized cRBCs. NB: Mature cRBCs can be conserved in SAGM medium (MacoPharma) at 4°C for up to one month.
- Day 18:
- Tests and representative results
The following tests are performed at the indicated days to verify proper cell proliferation and erythroid differentiation. Representative results presented on the figures were obtained using CD34+ cells isolated from leukapheresis.- Cells are counted every second day. See representative result of cell proliferation on Figure 2.
- May-Grünwald Giemsa morphology staining is done every second day using 0.05 x106 cells for Days 0 to 8 and 0.2x106 cells for the other days following instructions of the Giemsa stain manufacturer (Table 1). See representative results on Figure 3.
- Semisolid assays are performed in duplicate to measure hematopoietic progenitors every second day from Day 0 to Day 12 using 0.01x106 cells per assay following the instructions of the manufacturer of Methocult semi-solid medium (Table 1). See representative results on Figure 4.
- Analysis of the following cell markers is done every second day by FACS: CD34, CD36, CD71, GPA, LDS using 0.2 x106 cells per assay. See representative results on Figure 5.
- Hemoglobin is measured every second day by benzidine staining. See representative results on Figure 5.
- Gene expression is studied by RT-qPCR or microarray following RNA extraction from 1x106 cells. See representative results on Figure 6B.
Comments on the results:
Figure 1 outlines the protocol for ex vivo differentiation of human CD34+ cells to mature, hemoglobin-containing red blood cells. During erythroid differentiation, cells are highly proliferative as illustrated by the ~8,000-fold amplification shown on Figure 2 that was obtained using CD34+ cells isolated from leukapheresis. CD34+ cells isolated from bone marrow are slightly less proliferative with ~6,000-fold amplification while CD34+ cells isolated from cord blood can attain 50,000 fold amplification. Cells at particular stages of differentiation display recognizable sizes and morphologies that are illustrated on Figure 3. Notice the process of enucleation that occurs at the end of differentiation starting at Day 20. At Day 26, all cells have lost their nuclei (Figure 3).
Colony forming assays shown on Figure 4 are used to detect early and late hematopoietic progenitors during the first days of differentiation (Day 0 to Day 8). In addition to erythroid progenitors (CFU-E and BFU-E), we note that granulocyte-macrophage progenitors (CFU-GM) are significantly represented within the early CD34+ cells at Day 0. As differentiation proceeds, CFU-GM decrease significantly, and at Day 6 they have been completely replaced by erythroid progenitors BFU-E and CFU-E. It is also important to note that from Day 4 to Day 6, the early erythroid progenitors BFU-E are progressively replaced by their more differentiated counterparts CFU-E. By Day 10, erythroid cells have essentially lost their colony-forming capacity.
Finally, erythropoiesis is monitored by the expression of cell surface markers as shown in Figure 5. Note: 1) the progressive loss of the hematopoietic stem/progenitor marker CD34; 2) the progressive acquisition and loss of the progenitor marker CD36; 3) the progressive acquisition and loss of the erythroid progenitor and reticulocyte marker CD71; and 4) the increase of the erythroid precursor and erythrocyte marker GPA. Other markers of erythroid differentiation include hemoglobin synthesis starting at Day 8 (measured by benzidine staining-Figure 5) and cell enucleation starting at Day 20 as measured by the loss of the DNA stain LDS (Figure 5). Finally, erythroid differentiation can be measured by RT-qPCR of molecular markers such as the transcription factor TAL1 (Figure 6B) or the β-globin genes (data not shown).
Typically, 1 ml of cord blood / leukapheresis / bone marrow provides 0.35x105 / 0.5x105 / 1.1x105 CD34+ cells, which can theoretically produce 1.7x109 / 0.4x109 / 0.7x109 red blood cells with respective proliferation rates of 50,000 / 8,000 / 6,000. To obtain 50x106 red blood cells at the end of the differentiation procedure, you will need approximately 50 ml of culture medium.
Part II. Lentiviral mediated knockdown during ex vivo erythroid differentiation
Lentivirus is a tool of choice for gene transfer in human primary cells (see for example Amsellem et al. 9). Lentiviruses are often used to knockdown genes of interest in primary cells via delivery of targeted shRNA (see for example Laurenti et al. 10). In this protocol we target the transcription factor TAL1 in primary human erythroid cells. An shRNA with a scrambled sequence that does not correspond to any known gene is used as a negative control. GFP expression is used for evaluating transduction efficiency.
1. Preparation of lentiviral vector particles
To obtain lentiviral particles, human embryonic kidney 293T cells are cotransfected with three plasmids: one encompassing a second-generation virus packaging system coding for HIV-1 genes except env, vpr, vif, vpu and nef (i.e. psPAX2); one coding for the envelope of the vesicular stomatitis G-pseudotyped virus (i.e. pMD2G); and one lentiviral vector coding for the gene of interest (i.e. pBLOCK-it6-DEST(shTal1) coding for TAL1 shRNA, pBLOCK-it6-DEST(shScr) coding for scrambled shRNA or pWPIP coding for GFP) (Table 4). Lentiviral particles are collected from the cell culture supernatant of transfected 293T cells.
To avoid unwanted infection of cells, we perform all lentiviral experiments (preparation of viruses and infection) under a dedicated hood separate from other cell cultures. In addition, lentivirus-generated waste is discarded separately. Second generation lentiviruses described in this protocol are replication-incompetent and should be treated as Biosafety Level 2 organisms. Some institutions might have specific safety procedures regarding lentiviruses and we recommend that you follow established institutional guidelines.
Here we describe a protocol that will provide enough lentiviral particles to infect 1 to 2 millions primary erythroid cells at any given stage of differentiation. Volumes should be adjusted accordingly for scaling up or down. See Tables 4 and 5 for reagents.
- Transfection of 293T cells with calcium phosphate.
- One day before transfection, seed 13.2 x 106 293T cells in 20 ml of DMEM/High glucose medium containing 10% FBS and Pen/Strep in a
15-cm cell culture dish. NB. This should allow the cells to attain 70% confluency on the day of transfection. Best results are obtained using low
passage (<20) 293T cells.
Optional: dishes may be coated with type I collagen or Poly-L-lysine to enhance the adherence of 293T cells. - Two hours before transfection, aspirate off medium from 293T cells and gently add 20 ml of fresh medium preheated at 37°C. NB: We usually start the transfection at the end the day.
- Mix the following reagents in a 5 ml tube:
- 44.8 μg expression vector for your gene of interest (e.g. shRNA or GFP-see Table 4)
- 13.35 μg pMD2.G envelope vector.
- 33.48 μg psPAX2 packaging vector.
- Add enough sterile H2O to bring the volume to 1.116 ml.
- Add enough sterile H2O to bring the volume to 1.116 ml.
- Add 109.8 μl CaCl2 2.5M and vortex to mix.
- In a 50 ml conical tube, add 1.116 ml of 2xHBS buffer (Table 5).
- Under constant vortex agitation, add the DNA mixture dropwise into the 2xHBS buffer.
- Allow the precipitate to form at RT for 20 min. (no more than 30 min.).
- Add the 2.3 ml solution dropwise to the dish containing the 293T cells, swirl gently and transfer the cells to a 37°C incubator for 12-14 hours.
- Early the next morning, aspirate the medium and replace with 20 ml of fresh 37°C prewarmed medium.
- One day before transfection, seed 13.2 x 106 293T cells in 20 ml of DMEM/High glucose medium containing 10% FBS and Pen/Strep in a
15-cm cell culture dish. NB. This should allow the cells to attain 70% confluency on the day of transfection. Best results are obtained using low
passage (<20) 293T cells.
- Lentivirus harvest and concentration
- Forty-eight hours after transfection, harvest 20 ml medium containing the lentivirus and place in a 50 ml conical tube.
- Add 20 ml of 37°C prewarmed medium to the 293T cells and return to the incubator.
- Centrifuge the viral supernatant at 450 g for 10 min. to remove any cell debris and transfer into a new 50 ml tube. Alternatively the supernatant may be passed through a 0.45μm filter. Store the cleared supernatant at 4°C.
- Twenty-four hours later, harvest the remaining 20 ml of medium. NB: We find that the virus titers are roughly equivalent in both harvests i.e. 1x 106 TU/ml.
- To concentrate the viruses, viral supernatants are distributed into Beckman OptiSeal tubes (use 32.4 ml of supernatant per tube) and centrifuged at 100,000g for 1.5h at 4°C using a swinging rotor. N.B. Alternative methods to concentrate the virus supernatant include centrifugation at 47,000 g for 2h or at 3,000 g for 8h. Do not freeze the virus before concentration. Freezing will cause proteins to precipitate making it difficult to resuspend the virus.
- Immediately after ultracentrifugation, aspirate the supernatant slowly using a 10 ml plastic pipette and discard it. NB: Be careful not to disturb the pellet that can easily detach from the tube. After discarding the first 5ml of supernatant, we usually cut the top of the ultracentrifuge tube to help removing as much supernatant as possible. i.e. leave ~100-200 μl of medium at the bottom of the tube.
- Add 100 μl of 1xPBS, let the pellet dissolve at RT for 30 min. and resuspend by pipetting up and down 15-20 times. NB: Typically, we obtain ~200-300 μl of concentrated virus.
- The concentrated lentivirus preparation is titrated on NIH3T3 cells using a protocol from Invitrogen (Block-it lentiviral RNAi Expression System). NB: We typically obtain viral titers of 2 to 10x108 transducing units (TU)/ml.
- Virus can now be used for infection (see paragraph 2) or aliquoted and stored at -80°C for up to 6 months. NB: Do not submit virus suspensions to more than one freeze-thaw cycle. Viruses are stable for 4-5h at 37°C, 2-3 days at RT or one week at 4°C.
2. Lentiviral infection of primary erythroid cells for gene delivery
- Infection
Nucleated erythroid cells can be efficiently infected at any stage of differentiation as long as a high 50-100 MOI (multiplicity of infection) is used. Here we describe a typical experiment where lentiviruses expressing a TAL1 or scramble shRNA are used to infect erythroid cells at days 8 and 9 of differentiation using a MOI of 50 TU/cell. RNA is extracted every 24 hours to verify the level of TAL1 KD by RT-qPCR. GFP expression is measured in parallel by FACS for evaluating infection efficiency (see representative results on Figure 6).- Day 0:
- Prepare three 15 ml conical tubes containing 1x106 erythroid cells each (i.e. one tube for TAL1 knockdown, one tube for scrambled negative control, one tube for GFP expression control).
- Wash cells with 5 ml of supplemented IMDM medium containing SCF and EPO (see Part I), spin at 200 g for 5 min. and remove as much supernatant as possible.
- Add polybrene (Table 4) at 40 μg/ml final concentration into the concentrated virus solution.
- Add 100 μl of concentrated virus (i.e. 5x107 TU) containing polybrene to each tube of erythroid cells.
- Incubate for 1 h in the incubator at 37°C and mix by finger tapping every 10 min. N.B. infection should be carried out in minimal amounts of media to allow maximum adsorption of lentivirus onto the cells. In addition, mixing every 10 min. enables even distribution of virus across the cells.
- Add 900 μl supplemented IMDM medium containing SCF and EPO (Tables 2 and 3) in each tube.
- Transfer each sample in a 6-well plate and place in the 37°C incubator for 24 h.
- Day 1: Harvest cells in separate 15 ml tubes, spin them at 200 g for 5 min., remove the supernatant and repeat the infection (steps b to g above).
- Day 2: Wash the cells and resuspend them at 0.2x106 cell/ml into supplemented IMDM containing SCF and EPO and proceed as in Part I.
- Day 0:
- Representative results
Figure 6A illustrates the protocol for lentivirus infection of primary erythroid cells followed by RNA extraction. In one experiment, we have infected erythroid cells with lentivirus producing an shRNA targeted against the transcription factor TAL1 or a scrambled control 11. Figure 6B shows that we were successful in knocking down 75% of TAL1 transcripts in primary erythroid cells. Please also note the increase of TAL1 transcript level from Day 9 to Day 12 in the scrambled control underlying the importance of this transcription factor during erythroid differentiation. In another experiment, we have infected erythroid cells with lentivirus expressing the GFP protein. Figure 6C shows that three days after infection, most erythroid cells (87%) are expressing GFP as measured by fluorescence under microscope and FACS. This indicates a high efficiency of infection in primary erythroid cells. We have previously infected CD34+ cells at Day 0 with a similar efficiency of infection (data not shown). The most important parameter for efficient transduction of primary cells is to use a high MOI (i.e. concentrated lentiviruses). If a low MOI is used (i.e. non-concentrated virus), the efficiency of transduction will decrease.
3. Representative Results

Figure 1. Protocol outline for the isolation and ex vivo erythroid differentiation of human primary hematopoietic stem/progenitor cells. (Step1)
CD34+ cells (purity of 95 ± 3%) are isolated from cord blood, bone marrow or human peripheral blood mobilized with G-CSF (leukapheresis) using positive
immunomagnetic selection. (Step2) CD34+ cells are grown in liquid culture according to the indicated four-step protocol.
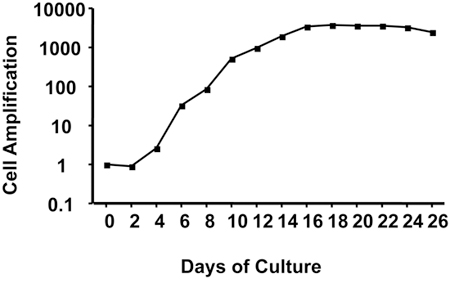
Figure 2. Cell amplification during ex vivo erythroid differentiation protocol. Results are in logarithmic scale. Representative results
obtained with CD34+ cells isolated from leukapheresis.

Figure 3. Cell morphological analysis during ex vivo erythroid differentiation. Cells are stained with May-Grünwald-Giemsa reagent at the indicated
time points during differentiation (magnification x 40). HSC: hematopoietic stem/progenitor cells; BFU-E: burst forming units –erythroid; CFU-E: colony forming
units –erythroid; Pro EB: pro-erythroblasts; Baso EB: basophilic erythroblasts; Poly EB: polychromatophilic erythroblasts; Ortho EB: orthochromatic erythroblasts;
RET: reticulocytes; RBC: red blood cells.
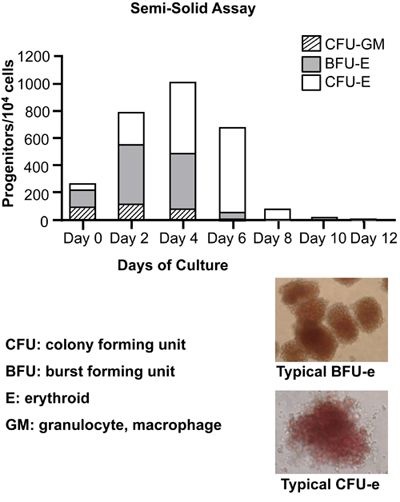
Figure 4. Colony-forming cell (CFC) assays to detect and quantify hematopoietic progenitors. At the indicated time points, 104 cells were
seeded in a semisolid methylcellulose medium in the presence of recombinant human cytokines: rh SCF, rh GM-CSF, rh G-CSF, rh Il-3, rh EPO to support optimal growth of
erythroid progenitors (BFU-E and CFU-E) and granulocyte-macrophage progenitors (CFU-GM). BFU: burst forming units; CFU: colony forming units. Typical picture of
BFU-E and CFU-E are also shown. Note the red-brown color due to hemoglobinization of BFU-E and CFU-E.
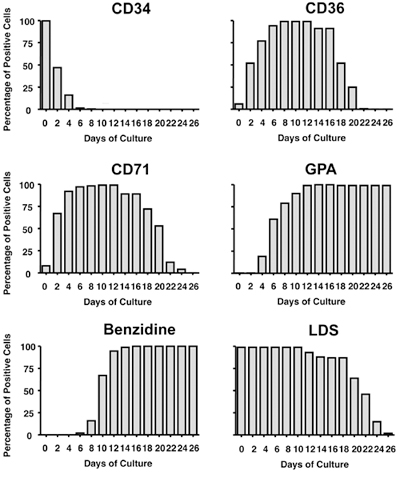
Figure 5. Kinetics of hematopoietic/erythroid markers during ex vivo erythroid differentiation. At the indicated time points, 2x105 cells were
harvested and analyzed for expression of indicated markers by FACS (CD34, CD36, CD71, GPA and LDS) or by microscopy (Benzidine). Bars represent percentages of positive
cells. CD34: transmembrane glycoprotein, present on human hematopoietic stem/progenitor cells; CD36: thrombospondin receptor, present on
progenitor cells; CD71: transferrin receptor, present on erythroid progenitors and reticulocytes; GPA: glycophorin A, present on
erythroid precursors and erythrocytes; Benzidine: hemoglobin stain; LDS: laser dye styryl, DNA stain.

Figure 6. Lentiviral-mediated gene transfer in human primary proerythroblasts. (A) Timeline for induction of TAL1 knockdown (KD) in primary
proerythroblasts using the lentiviral gene delivery system. (B) TAL1 knockdown was mediated by lentivirus-delivered anti-Tal1 shRNA (TAL1 KD)
or a scrambled control shRNA (Scr). Kinetics of TAL1 mRNA transcript level is measured by RT-qPCR. TAL1 transcript is expressed relative to 18S RNA. (C)
Lentivirus-delivered GFP was tested by microscopy (left panel) or FACS (right panel). 87% of cells were GFP positive.
4. Materials
| Reagent | Source | Catalog number |
| RPMI 1640 | Sigma | R8758 |
| Collagenase B | Roche Applied Science | 11088807001 |
| DNase I | Sigma | D5025-15KU |
| Pre-Separation Filters (30μm nylon mesh) | Miltenyi Biotec Inc | 130-041-407 |
| Ficoll-paque PLUS | GE Healthcare Bio-Sciences | 17-1440-03 |
| EasySep 10x Red Blood Cells Lysis Buffer | StemCell Technologies | 20120 |
| CD34 MicroBead Kit | Miltenyi Biotec Inc | 130-046-703 |
| LS Mini-Macs columns | Miltenyi Biotec Inc | 130-042-401 |
| autoMACS Running Buffer-MACS Separation Buffer | Miltenyi Biotec Inc | 130-091-221 |
| MiniMACS Separator | Miltenyi Biotec Inc | 130-042-102 |
| Mouse bone marrow stromal cells MS-5 (regularly test for mycoplasma contamination) | DSMZ | ACC 441 |
| Trypan Blue Solution (0.4%) | Sigma | T8154 |
| MEM Alpha+GlutaMAX | Gibco Invitrogen | 32571 |
| Trypsin 0.05%/EDTA | ThermoScientific | J101521 |
| May Grunwald Giemsa | Biostain Ready Reagents Ltd | RRSP54 |
| Methocult Colony-Forming Cell (CFC) Assays for Human Cells | StemCell Technologies | 04044 |
Table 1.
| Component | Stock solution | Volume 600 ml |
| IMDM | Sigma I3390 | 443.4 ml |
| Pen/Strept 1% (vol/vol) | Gibco 10378 | 6ml |
| L-glutamine 4x10-3M | 200mM (Gibco 25030) | 12 ml |
| Inositol 40μg/ml | 4mg/ml (Sigma I5125) preparation : Dissolve 28 mg powder in 7 ml IMDM and filter sterilize-stable for 1 week at 4°C. |
6ml |
| Folic acid 10μg/ml | 1 mg/ml (Sigma F7876) preparation : Dissolve 7 mg powder in 7 ml pre-warmed IMDM and filter sterilize-stable for 1 week at 4°C. Pre-warm before using. |
6ml |
| Monothioglycerol 1.6x10-4M | 0.16M (Sigma M6145) preparation : Dissolve 10 μl of 11.56M stock solution in 712 μl IMDM and filter sterilize-stable for 1 week at 4°C. |
0.6ml |
| Ferrous nitrate 90 ng/ml | 18μg/ml ( Sigma 8508) preparation: Dissolve 1.8 mg in 1ml distilled water. Take 0.1 ml, add 9.9 ml IMDM and filter sterilize-stable for 1 week at 4°C. |
3ml |
| Ferrous sulfate 900 ng/ml | 180 μg/ml ( Sigma F8633) preparation: Dissolve 18 mg in 1ml distilled water. Take 0.1 ml, add 9.9 ml IMDM and filter sterilize-stable for 1 week at 4°C. |
3ml |
| Bovine serum albumine-Insulin-Transferrin (BIT) 20% (vol/vol) |
StemCell Technologies 9500 | 120 ml |
Table 2.
| Component | Stock solution | Volume to add to 100 ml of supplemented IMDM (Table 2) |
| Hydrocortisone (HC) 10-6 M | 10-4 M (Sigma H2270) preparation : Dissolve 5mg powder in 1.03ml IMDM. Take 0.1 ml, add 9.9 ml IMDM and filter sterilize. Stable for 1 week at 4°C. |
1ml |
| Stem Cell Factor (SCF) 100ng/ml | 100 μg/ml (Cedarlane 4327-50) preparation: Dissolve 50 μg powder in 500 μl PBS+0.1% BSA and filter sterilize. Make aliquots of 100 μl and store at -20°C. |
100 μl |
| Erythropoietin (EPO) 3 IU/ml | 100 μg/ml equivalent of 12,000UI/ml (Feldan FB03-10-290) preparation: Dissolve 50 μg powder in 500 ml PBS+0.1% BSA and filter sterilize. Make aliquots of 25 ml and store at -20°C. |
25 μl |
| Interleukin 3 (IL3) 5 ng/ml | 100 μg/ml (Feldan FB03-10-330) preparation: Dissolve 10 μg powder in 100 μl PBS+0.1% BSA and filter sterilize. Make aliquots of 5 ml and store at -20°C. |
5 μl |
Table 3.
| Reagent | Source | Reference |
| 293T cells (regularly test for mycoplasma contamination) |
ATCC | CRL-11268 |
| DMEM/High Glucose | ThermoScientific | SH30243.01 |
| Hexadimethrine bromide (Polybrene) | Sigma | H9268-5G |
| OptiSeal ultracentrifugation tubes | Beckman Coulter | 361625 |
| psPAX2 plasmid | Addgene (D. Trono laboratory) | 12260 |
| pMD2.G plasmid | Addgene (D. Trono laboratory) | 12259 |
| pBLOCK-it6-DEST (shTal1) plasmid | Tal1 shRNA sequence cloned into the pBLOCK-it6-DEST plasmid from Invitrogen | Palii et al. EMBO J. |
| pBLOCK-it6-DEST (shScr) plasmid | Scrambled shRNA sequence cloned into the pBLOCK-it6-DEST plasmid from Invitrogen | Palii et al. EMBO J. |
| pWPI plasmid | Addgene (D. Trono laboratory) | 12254 |
Table 4.
| Buffer | Preparation | Final concentration |
| 2xHBS |
|
|
| CaCl2 |
|
|
| MS30 |
|
|
Table 5.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
A number of methods have been used previously to differentiate erythroid cells in culture with variable degrees of success. For example, some protocols without co-culture on MS-5 allows expansion of erythroid cells but are not efficient in producing fully mature enucleated red blood cells 12-17. While other methods allow efficient enucleation, it is at the expense of proliferation 18,19. The method we have used here, initially developed by the Douay laboratory 6, combines an initial liquid culture step, which prolongs cell amplification during early stages of erythroid differentiation and thus provides us with large numbers of early progenitors (necessary for genomics and proteomics studies), and a second step of co-culture on MS-5 mesenchymal cells, which is important to maximize hemoglobin synthesis and to obtain complete enucleation of red blood cells. Two limitations of our method are 1) the still insufficient degree of amplification at the earliest stages of differentiation, which so far has prevented us to fully characterize the very early erythroid progenitors at the protein level and 2) the high cost of performing this protocol, particularly on a large-scale.
The quality of cell culture reagents and cytokines is essential for ex vivo erythroid differentiation. Notably, the use of BIT (Table 2) as a serum replacement is critical. Indeed in our experience the use of BIT eliminated batch-to-batch variations that we experienced previously (likely due to differences in reagent purity) when using separate sources of Bovine serum albumin, Insulin and Transferrin. Another critical point is the quality of the MS-5 layer used for co-culture of erythroid cells. Indeed, MS-5 cells must be 70% confluent when erythroid cells are added and must be changed every 3-4 days. If MS-5 cells are too confluent, erythroid cells will not grow and differentiate properly. In addition, unhealthy MS-5 cells tend to detach, making the harvesting of erythroid cells difficult.
Previous methods have used retrovirus for gene-delivery in hematopoietic stem/progenitor cells in culture. However, retroviruses infect only dividing cells and are therefore not efficient in infecting the mainly quiescent CD34+ cells isolated from cord blood, bone marrow or leukapheresis 20. The main advantage of lentiviral approaches for gene-delivery is that it allows an efficient infection of cells independently of their proliferation status 7,21. Our lentiviral-mediated gene delivery allows one to efficiently knockdown or over-express specific transcription factors that can modify cell fate. Appropriate phenotypic analyses (cellular and molecular) can be devised after infection to reveal particular phenotypes. In addition, growth conditions may be altered to favor particular cell-types after infection. Along these lines, in this protocol we have chosen not to use any method to select lentivirus-infected cells for two main reasons: 1) our infection efficiency is very high (Figure 6C) and 2) we are concerned that the methods used to select lentivirus-infected cells (e.g. cell-sorting or antibiotic) are biased towards surviving/proliferating cells that are able to express either the antibiotic-resistance gene or the fluorescent marker at high levels thereby concealing a potential apoptotic phenotype.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank L. Douay and M.C Giarratana (Université Paris VI, France) for advice on the ex vivo erythroid differentiation, D. Allan and H. Atkins (OHRI, Canada) for providing blood samples (obtained under the Ottawa Hospital Research Ethics Board #2007804-01H), D. Trono (Ecole Polytechnique Federale de Lausanne) for providing the lentiviral vectors and F.J. Dilworth (OHRI, Canada) for critically reading the manuscript. This project was funded by a CIHR grant (MOP-82813) to M.B. C.G. P. is a recipient of an Ontario Research Fund Computational Regulomics Training Postdoctoral Fellowship. M.B. holds the Canada Research Chair in the Regulation of Gene Expression.
References
- Orkin, S. H., Zon, L. I. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 132, 631-644 (2008).
- Kim, S. I., Bresnick, E. H. Transcriptional control of erythropoiesis: emerging mechanisms and principles. Oncogene. 26, (2007).
- Friend, C., Patuleia, M. C., De Harven, E. Erythrocytic maturation in vitro of murine (Friend) virus-induced leukemic cells. Natl Cancer Inst Monogr. 22, 505-522 (1966).
- Weiss, M. J., Yu, C., Orkin, S. H. Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol Cell Biol. 17, 1642-1651 (1997).
- Lozzio, B. B., Lozzio, C. B., Bamberger, E. G., Feliu, A. S. A multipotential leukemia cell line (K-562) of human origin. Proc Soc Exp Biol Med. 166, 546-550 (1981).
- Giarratana, M. C. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat Biotechnol. 23, 69-74 (2005).
- Salmon, P., Trono, D. Production and titration of lentiviral vectors. Curr Protoc Neurosci. Chapter 4, (2006).
- Chao, M. P., Seita, J., Weissman, I. L. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harb Symp Quant Biol. 73, 439-449 (2008).
- Amsellem, S., Ravet, E., Fichelson, S., Pflumio, F., Dubart-Kupperschmitt, A. Maximal lentivirus-mediated gene transfer and sustained transgene expression in human hematopoietic primitive cells and their progeny. Mol Ther. 6, 673-677 (2002).
- Laurenti, E. Inducible gene and shRNA expression in resident hematopoietic stem cells in vivo. Stem Cells. 28, 1390-1398 (2010).
- Palii, C. G. Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J. 30, 494-509 (2011).
- Fibach, E., Manor, D., Oppenheim, A., Rachmilewitz, E. A. Proliferation and maturation of human erythroid progenitors in liquid culture. Blood. 73, 100-103 (1989).
- Wada, H. Expression of major blood group antigens on human erythroid cells in a two phase liquid culture system. Blood. 75, 505-511 (1990).
- Sui, X. Erythropoietin-independent erythrocyte production: signals through gp130 and c-kit dramatically promote erythropoiesis from human CD34+ cells. J Exp Med. 183, 837-845 (1996).
- Panzenbock, B., Bartunek, P., Mapara, M. Y., Zenke, M. Growth and differentiation of human stem cell factor/erythropoietin-dependent erythroid progenitor cells in vitro. Blood. 92, 3658-3668 (1998).
- Lindern, M. von The glucocorticoid receptor cooperates with the erythropoietin receptor and c-Kit to enhance and sustain proliferation of erythroid progenitors in vitro. Blood. 94, 550-559 (1999).
- Freyssinier, J. M. amplification and characterization of a population of human erythroid progenitors. Br J Haematol. 106, 912-922 (1999).
- Malik, P. An in vitro model of human red blood cell production from hematopoietic progenitor cells. Blood. 91, 2664-2671 (1998).
- Carlile, G. W., Smith, D. H., Wiedmann, M. Caspase-3 has a nonapoptotic function in erythroid maturation. Blood. 103, 4310-4316 (2004).
- Mazurier, F. Rapid analysis and efficient selection of human transduced primitive hematopoietic cells using the humanized S65T green fluorescent protein. Gene Ther. 5, 556-562 (1998).
- Salmon, P. High-level transgene expression in human hematopoietic progenitors and differentiated blood lineages after transduction with improved lentiviral vectors. Blood. 96, 3392-3398 (2000).
