Flow cytometry is a powerful tool allowing for the isolation and study of specific cell populations. This protocol describes steps for isolating LacZ-expressing cells from cochlear tissues from neonatal transgenic mice. Dissociated cochlear cells were labeled using fluorescent-conjugated substrates of β-galactosidase prior to separation via flow cytometry.
Method Article
Isolating LacZ-expressing Cells from Mouse Inner Ear Tissues using Flow Cytometry
Opens in a new tab
In This Article
Summary
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Abstract
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Isolation of specific cell types allows one to analyze rare cell populations such as stem/progenitor cells. Such an approach to studying inner ear tissues presents a unique challenge because of the paucity of cells of interest and few transgenic reporter mouse models. Here, we describe a protocol using fluorescence-conjugated probes to selectively label LacZ-positive cells from the neonatal cochleae.
The most common underlying pathology of sensorineural hearing loss is the irreversible damage and loss of cochlear sensory hair cells, which are required to transduce sound waves to neural impulses. Recent evidence suggests that the murine auditory and vestibular organs harbor stem/progenitor cells that may have regenerative potential1,2. These findings warrant further investigation, including identifying specific cell types with stem/progenitor cell characteristics. The Wnt signaling pathway has been demonstrated to play a critical role in maintaining stem/progenitor cell populations in several organ systems3-7. We have recently identified Wnt-responsive Axin2-expressing cells in the neonatal cochlea, but their function is largely unknown8.
To better understand the behavior of these Wnt-responsive cells in vitro, we have developed a method of isolating Axin2-expressing cells from cochleae of Axin2-LacZ reporter mice9. Using flow cytometry to isolate Axin2-LacZ positive cells from the neonatal cochleae, we could in turn execute a variety of experiments on live cells to interrogate their behavior as stem/progenitor cells. Here, we describe in detail the steps for the microdissection of neonatal cochlea, dissociation of these tissues, labeling of the LacZ-positive cells using a fluorogenic substrate, and cell sorting. Techniques for dissociating cochleae into single cells and isolating cochlear cells via flow cytometry have been described2,10-12. We have made modifications to these techniques to establish a novel protocol to isolate LacZ-expressing cells from the neonatal cochlea.
Protocol
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
1. Microdissection of the cochlea
- Prepare dissection microscope under hood by wiping surfaces with 70% ethanol and setting out ice blocks.
- Prepare 10-15 35 mm Petri dishes under sterile condition and fill half-way (~2 ml) with sterile Hank's Balanced Salt Solution with Calcium (HBSS).
- Prepare two 35 mm Petri dishes under sterile condition and fill half-way (~2 ml) with sterile 1x phosphate buffered solution (PBS).
- Sterilize micro-forceps in 50 ml conical Falcon tube with 70% ethanol.
- In a separate room from the sterile hood, use iris scissors to decapitate 12-15 postnatal (P) 0-3-day-old Axin2LacZ/+ reporter mice and place heads onto a 60 mm Petri dish on ice block.
- Using #5 Dumont forceps and iris scissors, excise the mandible and tongue and cut through the skull base at the level of the palate.
- Remove the overlying skin of the calvaria and transect the skull base by inserting the scissors at the inferior part of the foramen magnum to the middle of the palate.
- Make another incision with iris scissors from the cut edge of the palate to the superior aspect of the calvaria on both sides, being careful not to damage the otic capsule.
- Complete isolation of the temporal bone by making two 45-degree angled incisions starting from the superior aspect of the foramen magnum extending to the coronal cut at the hard palate.
- Place isolated temporal bones in 35 mm Petri dishes containing HBSS on ice block.
- Collect temporal bones from about 12-15 animals that have been sacrificed, for a total of 24-30 temporal bones.
- Transfer Petri dish with isolated temporal bones to previously prepared sterile dissection hood (1.1).
- Transfer four temporal bones to each pre-prepared 35 mm Petri dish containing HBSS (1.2) and place on ice block.
- Begin microdissection using two #55 forceps to gently remove the cortex, brain stem, and cerebellum in the temporal bone overlying the otic capsule.
- Without removing the otic capsule, chip away from parts of the cochlear capsule beginning at the entrance of the vestibulocochlear nerve into the otic capsule.
- Following exposure of the base of the cochlea, gently pull out the cochlea and surrounding tissues. Remove any remaining spiral ganglion and the stria vascularis beginning at the base.
- Transfer cochlea to 35 mm Petri dish containing PBS on ice block.
- For optimal tissue viability, the total time of dissection should be less than 60 min.
- Repeat above steps for 3-5 wild-type (WT) P0-P3 mice as controls for Axin2-LacZ mice.
2. Cell dissociation (modified from previously described methods)2,10
- Place 50 μl sterile PBS in the center of a well in a 6-well non-coated culture dish.
- Transfer up to 20 Axin2LacZ/+ cochleae into each 50 μl droplet of PBS. A new 50 μl droplet of PBS should be used for more than 20 cochleae.
- Pre-warm 50 μl aliquots of 0.125% trypsin for ~2 minutes in a 37°C water bath.
- Add 50 μl of pre-warmed 0.125% trypsin to PBS containing the cochleae.
- Place 6-well dish at 37°C for 8 min, taking care not to shake the dish.
- To terminate trypsinization, add 50 μl of soybean trypsin inhibitor to each 100 μl droplet.
- Add an additional 50 μl of Full Media (FM) (IGF-1, EGF, bFGF, HS, ampicillin in DMEM/F12)2
- Using a 300 μl blunt pipette tip with a p200 pipetteman set at 125 μl, gently triturate cells 80 times, being careful not to create bubbles.
- Following trituration of each droplet, add an additional 200 μl of FM to each well.
- Pass cells through a 40 μm blue Falcon cell strainer into a new well.
- Transfer cells to standard Falcon FACS tubes (approximately 300 μl remains following trituration and filtration).
- Split cells from wildtype cochleae into three tubes, diluting 100 μl with 300 μl FM in each tube. One tube will be used as a blank control, one will be stained with propidium iodide (PI, 1 μg/ml) only (not to be added at this step), while the last one will be stained with 3-carboxyumbelliferyl β-D-galactopyranoside (CUG, see 2.15) to detect background fluorescence.
- Place 400 μl of dissociated cells from Axin2-LacZ cochleae in each FACS tube. As a CUG only control, dilute 10 μl of cells in 390 μl FM (see tube F in Step 2.15). Bring volume of any remaining tubes to 400 μl.
- Incubate cells in a 37°C humidified incubator for 10 min.
- Prepare CUG solution:
- Prepare 200 μl of CUG for each FACS tube requiring staining.
- Dilute CUG in FM 1:50 in a dark hood with minimum light exposure.
- Pre-warm diluted CUG at 37°C for 3-5 minutes.
- Next, add 200 μl of diluted CUG to each appropriate FACS tube. For tubes not designated to have CUG, add FM only (see 2.15).
- At this juncture, there should be five categories of tubes with the following contents (do not add PI at this step):
- Wildtype - without CUG or PI (for compensation, see step 3.5-6)
- Wildtype - with PI only (for compensation)
- Wildtype - with CUG and PI (for control against Axin2-LacZ cells)
- Axin2-LacZ - with CUG only (for compensation)
- Axin2-LacZ - with CUG and PI (for actual cell sorting)
- Axin2-LacZ - with CUG and PI (for event analysis)
- Protect all tubes from light and incubate them in a 37°C humidified incubator for 25 minutes.
- Prepare fresh HEPES + Fetal Bovine Serum (FBS) solution for halting reaction following incubation:
- Dilute 400 μl of freshly thawed FBS in 10 ml of sterile 10 mM HEPES (in PBS) in a 15 ml Falcon tube.
- Place tube on ice until completion of 2.16.
- Add 1.8 ml of ice-cold HEPES solution to each FACS tube under a sterile hood with lights off to minimize light exposure.
- Centrifuge cells at 1200rpm for 5 minutes.
- Remove supernatant and re-suspend cells in 300 μl FM, pool all aliquots for the sorting sample (Axin2-LacZ stained with CUG and PI).
- Add PI to each tube to achieve a final concentration of 1 μg/ml.
- Place cells on ice and protect from light.
3. Flow Cytometry
- We use a BD Aria II equipped with 488-nm, 633-nm, and 407-nm lasers. Detection of CUG was done using the 407-nm violet laser and PI using the 488-nm blue laser at the Stanford Shared FACS Facility.
- A trained flow cytometer operator is required. Proceed to the next step after warming up the lasers, calibrating the cytometer detectors using manufacturer instructions, and setting optimal sorting parameters. In our experience, we found the use of a 100 μm nozzle optimizes cell viability for subsequent experiments.
- The following parameters were used: threshold at 5,000 events, flow rate less than 3,000 events/second, forward scatter (FSC) at 125 volts and side scatter (SSC) at 250 volts, PE-Cy5 channel was used for PI and Cascade Blue for CUG.
- Flush cytometer chamber with 70% ethanol and PBS twice each in that order.
- Begin analysis with unstained, wildtype cells (tube A in Step 2.15). Collect 1,000 events to determine cell distribution on the FSC versus SSC plot. If scatter is not desired, then adjust FSC and SSC parameters.
- Perform compensation using unstained WT (tube A), PI-stained WT (tube B), and CUG-stained Axin2-LacZ (tube D) cells. Compensation will allow for more accurate signal discrimination among the different laser wavelengths.
- Start analysis with wildtype PI- and CUG-stained cells (tube C) and set gating parameters for:
- Exclusion of debris based on FSC versus SSC plot.
- Exclusion of doublets and clumps using FSC-height versus FSC-area plot.
- Exclusion of dead cells based on FSC-area versus PE-Cy5-area plot.
- Set up plot of FSC-area versus Cascade Blue-area with any gate placement.
- Next, analyze 10,000 events from sort sample tube that contains Axin2-LacZ PI and CUG stained cells (tube F).
- All gates set using wildtype cells should be applied here.
- Using plots for the wildtype cells, create gates for Cascade Blue-high cells that is less than 1% of the total number of events.
- This same gates of Cascade Blue-high now encompass approximately 15-20% of cells in the Axin2-LacZ sample, making these Axin2-high cells.
- Using the Axin2-LacZ cells' analysis plot, create a gate for the least fluorogenic 15-20% of cells, henceforth Axin2-low cells.
- Based on the aforementioned gating strategy, sort cells into FACS tubes containing 500 μl FM, one tube for Axin2-high and the other for Axin2-low cells. Make sure collection tubes are in a chamber cooled to 4°C during the sort.
- Record number of cells counted by flow cytometer for each population.
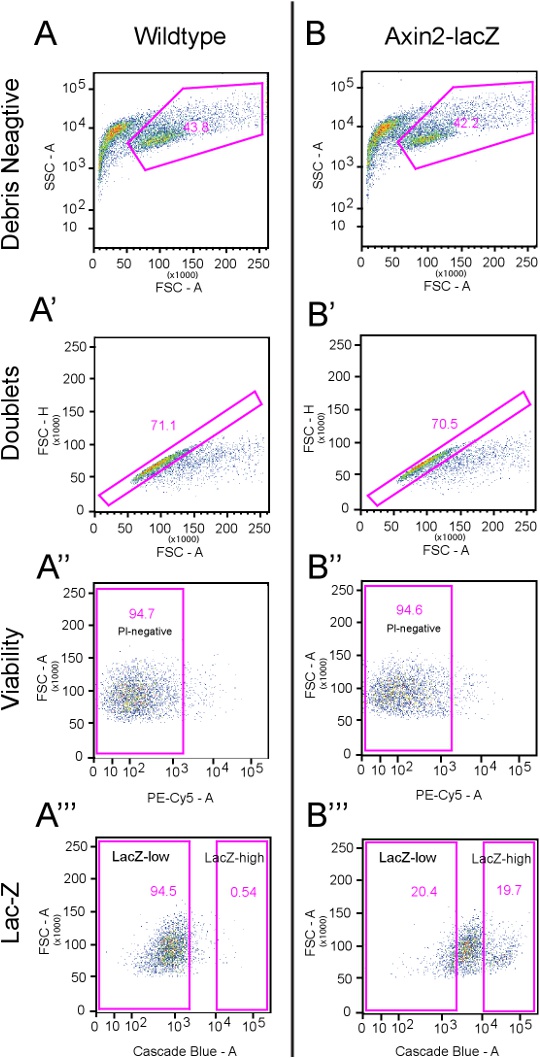
4. Representative Results (see Figure 2):
With the above protocol, we typically recover 40-45% debris-free cells from both the wildtype and Axin2LacZ/+ cochleae (Figures 2A and B), with about 70% of them free of doublets (see Figures 2A' and B'). Most sorted cells are viable and did not take up PI (~95%) (Figures 2A'' and B''). On average, the top ~20% CUG-positive cells are considered Axin2-LacZ-positive cells (Figure 2B''') and correlate to about 15,000 Axin2-LacZ-high cells per 10-12 animals.
Flow cytometry re-sort analysis of Axin2-high cells revealed greater than 93% purity in the Cascade Blue channel. In order to further confirm the identity of these cells, we performed post-sort immunostaining of both Axin2-high and Axin2-low cells using anti-β-galactosidase antibodies, and found that the Axin2-high cells contained ~93.4% β- galactosidase-positive cells (greater than 1,500 cells analyzed) and that Axin2-low cells contained ~4.8% β-galactosidase-positive cells.
Figures and Tables:

Figure 1.Experimental paradigm. The above figure depicts the overall experimental strategy and techniques as described through the protocol section. Cochleae from wildtype and Axin2LacZ/+ mice are harvested and dissociated into single cells, which were then treated with CUG, a fluorescent substrate for β-galactosidase. Axin2-LacZ-high and -low cells were then separated out via flow cytometry. Click here to view a full-sized version of this image.
{kind=link}

Figure 2.Gating Strategies. This figure illustrates the flow cytometry gating strategies used. A) Dissociated wildtype cells that underwent the same treatment as Axin2-LacZ cells are analyzed here using a total of 10,000 events. The first panel demonstrates the side scatter area (SSC-A) versus forward scatter area (FSC-A). Here, a distinct group of events are of a much larger size than those events closer to the y-axis. These events are designated as the "Debris Negative." The next gate, A', analyzes the "Debris Negative" cells from panel A, which is 43.8% of the total number of events (see table below). In this panel, the forward scatter height (FSC-H) versus the FSC-A is used to exclude doublets. Events deviating from the expected linear nature of the plot are excluded. This preparation typically yields ~70% events within the designated gate, and the remaining cells are excluded. In panel A'', propidium iodide (PI) as detected by the PE-Cy5 channel, is used to label dead cells (5.3%). In the next panel (A'''), the Cascade blue channel is used to detect the fluorescent substrate CUG. Here, the LacZ-high gates include less than 1% of total events. B) Dissociated cells from Axin2-LacZ mice treated with CUG and PI are analyzed here using a total of 10,000 events. Identical gates from panel A are applied here. B' shows that 29.5% of cells are excluded as doublets while B'' demonstrates that 5.4% are not viable. The initial gates for LacZ-high and -low cells are drawn using panel B''' with LacZ-high initially set at 19.7% and LacZ-low at 20.4%. Click here to view a full-sized version of this image.
{kind=link}
| Event number | Percentage | ||
| A | Wildtype Analysis | 10,000 | |
| A' | Debris Negative | 4,381 | 43.80% |
| A'' | Doublet Negative | 3,116 | 71.10% |
| A''' | PI Negative | 2,951 | 94.70% |
| LacZ-high | 16 | 0.54% | |
| LacZ-low | 2,790 | 94.50% | |
| B | Axin2-LacZ Analysis | 10,000 | |
| B' | Debris Negative | 4,218 | 42.2% |
| B'' | Doublet Negative | 2,973 | 70.50% |
| B''' | PI Negative | 2,813 | 94.6% |
| LacZ-high | 553 | 19.70% | |
| LacZ-low | 573 | 20.40% |
Table 1. Quantitative analyses of gating strategies in Figure 2.
Access restricted. Please log in or start a trial to view this content.
Discussion
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Independent research studies have characterized limited regenerative capacity within the neonatal cochleae2,10,12,13. Using a GFP-reporter mice and flow cytometry, White and colleagues isolated specific cochlear supporting cells and found them to have progenitor cell characteristics12.
The canonical Wnt pathway has been demonstrated to mark stem/progenitor cell populations in multiple organ systems including the brain, mammary gland, hematopoietic system, skin, and gastro...
Access restricted. Please log in or start a trial to view this content.
Disclosures
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
No conflicts of interest declared.
Acknowledgements
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
We thank S. Heller, K. Oshima, R. Nusse, and Y. Zeng for fruitful discussions and C. Tang, A. Lee, E. Liaw, and the Stanford Shared FACS Facility staff for technical assistance. This work was supported by Howard Hughes Medical Institute Medical Research Training Fellowship, Stanford University Medical Scholars program (both to T.A.J.), Stanford University Dean's Fellowship (to R.C.), American Otological Society, Triological Society, Percy Memorial Award, the Akiko Yamazaki and Jerry Yang Faculty Scholar Fund, and NIDCD/NIH K08 DC011043 (all to A.G.C.).
Access restricted. Please log in or start a trial to view this content.
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| Petri dish, polystyrene, sterile Geriner Bio-one 35 x 10 mm | VWR international | 82050-540 | |
| BD Falcon Cell Strainers, Sterile, BD Biosciences, blue, 40 μm | VWR international | 21008-949 | |
| Hanks’ Balanced Salt Solution (HBSS). Solution with Calcium, Magnesium, and without Phenol Red, Sterile | VWR international | 45000-456 | |
| Cellstar centrifuge tubes, polypropylene, sterile, greiner bio-one, 50 ml | VWR international | 82050-346 | |
| Cellstar centrifuge tubes, polypropylene, sterile, greiner bio-one, 15 ml | VWR international | 82050-278 | |
| B-27 Serum-Free Supplement (50x), liquid | Invitrogen | 17504-044 | |
| N-2 Supplement (100x), liquid | Invitrogen | 17502-048 | |
| bFGF, fibroblast growth factor-basic human | Sigma-Aldrich | F0291 | |
| IGF-1, insulin-like growth factor-1 from mouse | Sigma-Aldrich | I8779 | |
| EGF, epidermal growth factor human | Sigma-Aldrich | E9644 | |
| Dulbecco’s Modified Eagle’s Medium/Ham’s F-12 50/50 Mix: 1X, with L-Glutamine and 15 mM HEPES | VWR international | 45000-350 | |
| BD Falcon Round-Bottom Tubes, Disposable, Polystyrene, 12x75 | VWR international | 60819-295 | |
| Trypsin, 0.25% (1X) with EDTA 4Na, liquid | Invitrogen | 25200-056 | |
| ep Dualfilter TIPS 300uL, 960 TIPS | Eppendorf | 22491245 | |
| Trypsin Inhibitor, Soybean, Purified | Worthington Biochemical | LS003570 | |
| BD Falcon 40um Cell Strainers | BD Biosciences | 21008-949 | |
| Multiwell Plates, Polystyrene, Greiner Bio-One, Nontreated Plates, 6 wells | VWR international | 82050-846 | |
| Marker Gene FACS Blue LacZ beta-Galactosidase Detection Kit | Marker Gene Technologies | M0255 |
References
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
- Li, H., Liu, H., Heller, S. Pluripotent stem cells from the adult mouse inner ear. Nat. Med.. 9, 1293-1299 (2003).
- Oshima, K. Differential distribution of stem cells in the auditory and vestibular organs of the inner ear. J. Assoc. Res. Otolaryngol. 8, 18-31 (2007).
- Barker, N. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 449, 1003-1007 (2007).
- Jaks, V. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet. 40, 1291-1299 (2008).
- Kalani, M. Y. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 105, 16970-16975 (2008).
- Willert, K. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 423, 448-452 (2003).
- Zeng, Y. A., Nusse, R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell. 6, 568-577 Forthcoming.
- Chai, R. Dynamic Expression of Lgr5, a Wnt Target Gene, in the Developing and Mature Mouse Cochlea. J. Assoc. Res. Otolaryngol. , Forthcoming.
- Lustig, B. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell Biol. 22, 1184-1193 (2002).
- Diensthuber, M., Oshima, K., Heller, S. Stem/progenitor cells derived from the cochlear sensory epithelium give rise to spheres with distinct morphologies and features. J. Assoc. Res. Otolaryngol. 10, 173-190 (2009).
- Savary, E. Cochlear stem/progenitor cells from a postnatal cochlea respond to Jagged1 and demonstrate that notch signaling promotes sphere formation and sensory potential. Mech. Dev. 125, 674-686 (2008).
- White, P. M., Doetzlhofer, A., Lee, Y. S., Groves, A. K., Segil, N. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature. 441, 984-987 (2006).
- Zhang, Y. Isolation, growth and differentiation of hair cell progenitors from the newborn rat cochlear greater epithelial ridge. J. Neurosci. Methods. 164, 271-279 (2007).
Access restricted. Please log in or start a trial to view this content.
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request Permission