

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Cet article décrit les protocoles de préparation des échantillons, de réduction des données et d’analyse des données dans les études d’écho de spin neutronique (NSE) des membranes lipidiques. Le étiquetage judicieux des lipides au deutérium permet d’accéder à différentes dynamiques membranaires sur des échelles de longueur et de temps mésoscopiques, sur lesquelles se produisent des processus biologiques vitaux.
Abstract
Les bicouches lipidiques forment la matrice principale des membranes cellulaires et constituent la principale plate-forme pour l’échange de nutriments, les interactions protéine-membrane et le bourgeonnement viral, entre autres processus cellulaires vitaux. Pour une activité biologique efficace, les membranes cellulaires doivent être suffisamment rigides pour maintenir l’intégrité de la cellule et de ses compartiments, mais suffisamment fluides pour permettre aux composants membranaires, tels que les protéines et les domaines fonctionnels, de diffuser et d’interagir. Cet équilibre délicat des propriétés des membranes élastiques et fluides, et leur impact sur la fonction biologique, nécessitent une meilleure compréhension de la dynamique collective des membranes sur les échelles de longueur et de temps mésoscopiques des processus biologiques clés, par exemple les déformations membranaires et les événements de liaison aux protéines. Parmi les techniques qui peuvent sonder efficacement cette plage dynamique, il y a la spectroscopie par écho de spin neutronique (NSE). Combiné au labelage au deutérium, le NSE peut être utilisé pour accéder directement aux fluctuations de flexion et d’épaisseur ainsi qu’à la dynamique mésoscopique de certaines caractéristiques de la membrane. Cet article fournit une brève description de la technique NSE et décrit les procédures pour effectuer des expériences NSE sur les membranes liposomales, y compris des détails sur les schémas de préparation et de deutération des échantillons, ainsi que des instructions pour la collecte et la réduction des données. L’article présente également des méthodes d’analyse de données utilisées pour extraire des paramètres clés de la membrane, tels que le module de rigidité de flexion, le module de compressibilité de surface et la viscosité dans le plan. Pour illustrer l’importance biologique des études NSE, certains exemples de phénomènes membranaires sondés par NSE sont discutés, à savoir l’effet des additifs sur la rigidité de flexion de la membrane, l’impact de la formation de domaine sur les fluctuations membranaires et la signature dynamique des interactions membrane-protéine.
Introduction
La compréhension des membranes cellulaires et de leur fonction a remarquablement évolué au cours des dernières décennies. L’ancienne vision des membranes cellulaires en tant que bicouches lipidiques passives qui définissent les limites cellulaires et abritent les protéines membranaires1 s’est progressivement transformée en un modèle dynamique dans lequel les bicouches lipidiques jouent un rôle important dans la régulation des processus biologiques vitaux, y compris la signalisation cellulaire, l’échange moléculaire et la fonction protéique – pour n’en nommer que quelques-uns2,3,4,5,6. Cette prise de conscience que les membranes cellulaires sont très dynamiques, en constante évolution et redistribution moléculaire, a poussé les explorations scientifiques au-delà des structures d’équilibre des membranes7,8,9. En conséquence, de multiples approches ont été développées pour étudier les différents modes dynamiques dans les membranes lipidiques biologiques et bioinspirées. À ce jour, la majorité de ces études se sont principalement concentrées sur les mouvements moléculaires diffusifs10,11,12,13 et les fluctuations de forme macroscopiques14,15,16, laissant une lacune significative dans la compréhension de la dynamique membranaire intermédiaire, c’est-à-dire les fluctuations collectives des assemblages lipidiques constitués de quelques 10 à 100 molécules lipidiques. Ces dynamiques se produisent sur des échelles de longueur de quelques dizaines à quelques 100 Å et sur des échelles de temps de sous-ns à quelques centaines de ns (voir figure 1),appelées ici échelles mésoscopiques. C’est en effet à ces échelles que l’activité biologique clé a lieu au niveau de la membrane17. Cela inclut le bourgeonnement viral18, le canal gating19et les interactions membrane-protéine20. Il est également important de souligner que le paysage énergétique des protéines membranaires21,22 montre que les changements conformationnels dans les protéines – nécessaires à leur rôle régulateur – se produisent sur les échelles de temps ns23 des fluctuations membranaires collectives, soulignant davantage l’importance de la dynamique mésoscopique dans la fonction biologique des membranes cellulaires et de leurs analogues bioinspirés20. Cet article se concentre sur les deux principaux modes dynamiques mésoscopiques dans les membranes lipidiques, à savoir les fluctuations de flexion et les fluctuations d’épaisseur.
Le principal défi pour sonder directement ces modes de fluctuation est la difficulté d’accéder simultanément à leurs échelles spatiales et temporelles à l’aide de méthodes de spectroscopie standard. L’autre défi est que les techniques de contact direct pourraient avoir un impact sur les mêmes fluctuations qu’elles sont censées mesurer16. Ceci est encore exacerbé par la complexité compositionnelle et structurelle des membranes biologiques24,25, qui se traduit par des caractéristiques membranaires non homogènes, y compris la formation de domaine lipidique26,27,28,29,30 et l’asymétrie membranaire31,32,33- exigeant des sondes sélectives pour comprendre la dynamique des différentes caractéristiques de la membrane. Heureusement, ces défis peuvent être surmontés avec des méthodes de spectroscopie neutronique non invasives, telles que l’écho de spin neutronique (NSE), qui accèdent intrinsèquement aux échelles de longueur et de temps requises, et permettent d’autres études des caractéristiques sélectives de la membrane sans modifier leur environnement physico-chimique34. En effet, au cours des dernières années, la spectroscopie NSE a évolué pour devenir une sonde unique et puissante de dynamique membranaire collective35. Les résultats des études NSE sur les membranes lipidiques ont produit de nouvelles connaissances sur les propriétés mécaniques36,37 et viscoélastiques38,39 des membranes lipidiques et ont jeté un nouvel éclairage sur leur rôle potentiel dans la fonction biologique40,41.
La technique de spectroscopie NSE est basée sur une conception d’instrument interférométrique, proposée pour la première fois par Mezei42,utilisant une série de spin-flippers et de bobines magnétiques pour contrôler la précession du spin neutronique lorsque les neutrons traversent l’instrument. La conception repose sur la mise en miroir magnétique des éléments du champ magnétique par rapport à la position de l’échantillon(Figure 1A). Cela implique qu’en l’absence d’échange d’énergie entre le neutron et l’échantillon, le neutron effectue le même nombre de précessions de spin, dans des directions opposées, dans la première et la deuxième moitié de l’instrument (remarquez le π-flipper entre les deux bobines de précession). En conséquence, l’état de spin final du neutron reste inchangé par rapport à l’état initial - un phénomène appelé écho de spin (voir neutron transparent à la figure 1A). Cependant, lorsque le neutron interagit énergétiquement avec l’échantillon, l’échange d’énergie modifie le nombre de précessions de spin dans la seconde moitié de l’instrument, conduisant à un état de spin final différent (voir Figure 1A). Ceci est détecté expérimentalement comme une perte de polarisation, comme nous le verrons plus loin dans cet article. Pour plus de détails sur la technique NSE, le lecteur est référé aux documents techniques dédiés42,43,44,45.
Ici, une description simplifiée est présentée pour fournir une estimation approximative de la durée et des échelles de temps accessibles avec NSE. Les échelles de longueur sont déterminées par la gamme des transferts de vecteurs d’onde réalisables, Q = 4π sin θ/λ, où 2θ est l’angle de diffusion et λ est la longueur d’onde du neutron. On peut voir que Q est défini par la gamme de longueurs d’onde et l’étendue de rotation du deuxième bras du spectromètre (voir Figure 1A). Une plage Qtypique sur les spectromètres NSE est ~0,02-2 Å-146,47, et jusqu’à 0,01-4 Å-1 avec des mises à niveau récentes48,49, correspondant à des échelles spatiales de ~ 1-600 Å. D’autre part, l’échelle de temps accessible est calculée à partir de l’angle total de précession (ou phase) acquis par le neutron à l’intérieur des bobines de précession magnétique, et se trouve êtrede 50:  . Dans cette expression, t est le temps de Fourier défini comme
. Dans cette expression, t est le temps de Fourier défini comme  , où est le rapport
, où est le rapport  gyromagnétique des neutrons,
gyromagnétique des neutrons,  est la longueur de la bobine et est la force du champ magnétique de
est la longueur de la bobine et est la force du champ magnétique de  la bobine. Il convient de souligner que le temps de Fourier est une quantité qui dépend strictement de la géométrie de l’instrument, de l’intensité du champ magnétique et de la longueur d’onde des neutrons. Par exemple, en utilisant des neutrons de longueur d’onde
la bobine. Il convient de souligner que le temps de Fourier est une quantité qui dépend strictement de la géométrie de l’instrument, de l’intensité du champ magnétique et de la longueur d’onde des neutrons. Par exemple, en utilisant des neutrons de longueur d’onde  = 8 Å et des réglages d’instrument de = 1,2 m et = 0,4 T, le temps de Fourier est calculé à t ~ 50 ns. Expérimentalement, le temps de Fourier est réglé en modifiant le courant dans les bobines de précession (c’est-à-dire l’intensité du champ magnétique) ou en utilisant différentes longueurs d’onde de neutrons, ce qui donne des échelles de temps NSE typiques de ~ 1 ps à 100 ns. Cependant, les récentes mises à niveau des spectromètres NSE ont permis d’accéder à des temps de Fourier plus longs, jusqu’à ~400 ns sur le spectromètre J-NSE-Phoenix au Heinz Maier-Leibnitz Zentrum51 et le spectromètre SNS-NSE au Oak Ridge National Lab48,et jusqu’à ~1 000 ns au spectromètre IN15 NSE à l’Institut Laue-Langevin (ILL)49.
= 8 Å et des réglages d’instrument de = 1,2 m et = 0,4 T, le temps de Fourier est calculé à t ~ 50 ns. Expérimentalement, le temps de Fourier est réglé en modifiant le courant dans les bobines de précession (c’est-à-dire l’intensité du champ magnétique) ou en utilisant différentes longueurs d’onde de neutrons, ce qui donne des échelles de temps NSE typiques de ~ 1 ps à 100 ns. Cependant, les récentes mises à niveau des spectromètres NSE ont permis d’accéder à des temps de Fourier plus longs, jusqu’à ~400 ns sur le spectromètre J-NSE-Phoenix au Heinz Maier-Leibnitz Zentrum51 et le spectromètre SNS-NSE au Oak Ridge National Lab48,et jusqu’à ~1 000 ns au spectromètre IN15 NSE à l’Institut Laue-Langevin (ILL)49.
Outre l’accès direct à la durée et à l’échelle de temps de la dynamique membranaire, NSE a les capacités inhérentes de la sensibilité isotopiqueneutronique 52. Plus précisément, la capacité des neutrons à interagir différemment avec les isotopes de l’hydrogène, l’élément le plus abondant dans les systèmes biologiques, entraîne une densité de longueur de diffusion des neutrons différente,34 ou NSLD (l’équivalent de l’indice optique de réfraction50),lorsque le protium est substitué par du deutérium. Cela permet une approche connue sous le nom de variation de contraste, qui est couramment utilisée pour mettre en évidence des caractéristiques spécifiques de la membrane ou en dissimuler d’autres – ce dernier scénario est appelé correspondance de contraste. Une application fréquente de variation/correspondance de contraste est la substitution de l’eau (NSLD = -0,56 × 10-6 Å-2) par de l’eau lourde ouD2O (NSLD = 6,4 × 10-6 Å-2) pour amplifier le signal neutronique des membranes lipidiques protiées (NSLD ~ 0 × 10-6 Å-2). Cette approche est très efficace dans les études de la structure membranaire car la pénétration de D2O dans la région du groupe de tête de la membrane permet de déterminer avec précision les épaisseurs de membrane (voir Figure 2A,panneau de gauche) et de l’emplacement des différents sous-groupes lipidiques lorsque des modèles plus sophistiqués sont appliqués53,54. Cet article met en évidence quelques exemples sur l’utilisation de la variation de contraste pour les études de la dynamique collective dans les membranes biomimétiques et certaines caractéristiques membranaires.
Ici, l’efficacité de NSE à fournir des informations uniques sur les propriétés dynamiques et fonctionnelles des membranes est illustrée par des exemples tangibles d’études NSE sur des systèmes de membranes lipidiques modèles et biologiquement pertinents en mettant l’accent sur la dynamique à méso-échelle dans les membranes autoportantes, sous la forme de suspensions liposomales. Pour les mesures NSE de la dynamique des membranes dans le plan, le lecteur est référé à des publications dédiées sur la spectroscopie d’écho-spin neutronique d’incidence de pâturage (GINSES)55,56 et d’autres études de piles de membranes multilamellaires alignées57,58,59,60.
Pour simplifier, cet article met en évidence trois schémas différents de deutération membranaire illustrés sur un système de bicouche lipidique bien étudié formant ou séparant les phases de mélanges de 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) et de 1,2-distéaroyl-sn-glycero-3-phosphocholine (DSPC)61,62. Les deux lipides sont caractérisés par un décalage dans leur longueur de chaîne hydrocarbonée (14 carbones/queue en DMPC vs 18 carbones/queue en DSPC) et leur température de transition gel-fluide (Tm, DMPC = 23 °C vs Tm, DSPC = 55 °C). Il en résulte une séparation latérale de phase dans les membranes DMPC:DSPC à des températures comprises entre les températures de transition supérieures et inférieures du mélange63. Les schémas de deutération considérés ici sont choisis pour démontrer les différents modes dynamiques accessibles dans les mesures NSE sur les membranes liposomales, à savoir les fluctuations de flexion, les fluctuations d’épaisseur et les fluctuations sélectives de flexion / épaisseur des domaines latéraux. Toutes les compositions lipidiques sont rapportées pour les bicouches DMPC:DSPC préparées à une fraction molaire de 70:30, en utilisant des variantes protiées et perdeuterées de DMPC et DSPC disponibles dans le commerce. Toutes les étapes de préparation de l’échantillon sont basées sur 4 mL de suspension liposomale, en D2O, avec une concentration lipidique de 50 mg/mL, pour une masse lipidique totale de Mtot = 200 mg par échantillon.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Schéma de deutération requis pour l’expérience
- Pour les mesures de fluctuation de flexion, faire des liposomes entièrement protiés dans D2O (D 99,9%) ou D2O-tampon (par exemple, tampon phosphate préparé avec D2O au lieu de H2O). Utilisez du DMPC entièrement protié (C36H72NO8P) et du DSPC (C44H88NO8P) avec
 133,4 mg, où XDMPC et XDSPC sont les fractions molaires de DMPC et DSPC, ici réglés à 0,7 et 0,3, respectivement, et MwDMPC et MwDSPC sont les poids molaires donnés par 677,9 g/mol et 790,1 g/mol, respectivement. De même, mDSPC = 66,6 mg. Ce schéma de deutération augmente le contraste de diffusion entre la membrane (NSLD ~ 0 × 10-6 Å -2) et le tampon deutéré (NSLD ~ 6,4 × 10-6 Å -2) et amplifie le signal des ondulations membranaires (voir le panneau de gauche de la figure 2A).
133,4 mg, où XDMPC et XDSPC sont les fractions molaires de DMPC et DSPC, ici réglés à 0,7 et 0,3, respectivement, et MwDMPC et MwDSPC sont les poids molaires donnés par 677,9 g/mol et 790,1 g/mol, respectivement. De même, mDSPC = 66,6 mg. Ce schéma de deutération augmente le contraste de diffusion entre la membrane (NSLD ~ 0 × 10-6 Å -2) et le tampon deutéré (NSLD ~ 6,4 × 10-6 Å -2) et amplifie le signal des ondulations membranaires (voir le panneau de gauche de la figure 2A). - Pour mesurer la dynamique de flexion de certaines caractéristiques de la membrane latérale, par exemple la dynamique de la matrice dans les membranes DMPC:DSPC à séparation de phase, utilisez du DMPC protié (C36H72NO8P) et deutéré, DSPC-d83 (C44H5NO883, Mw 873,7 g/mol), de telle sorte que mDMPC = 128,8 mg et mDSPC-d83 = 71,2 mg. Ce schéma de deutération minimise la diffusion à partir des domaines riches en DSPC indésirables, ce qui permet de mesurer sélectivement les fluctuations de flexion de la matrice riche en DMPC (voir la figure 2B au milieu).
REMARQUE: Pour trouver la deutération lipidique optimale requise pour un schéma d’appariement de contraste spécifique, utilisez les calculateurs de densité de longueur de diffusion (SLD) disponibles sur le Web, tels que celui développé par le NIST Center for Neutron Research64. Ces interfaces web sont équipées d’outils conviviaux pour faciliter le calcul du SLD des lipides avec différents degrés de deutération, ainsi que celui des mélanges lipidiques. - Pour les mesures NSE des fluctuations moyennes de l’épaisseur de la membrane (sans contraste latéral), utiliser des variantes deutérées par la queue des lipides constitutifs, à dire DMPC-d54 (C36H18NO854, 732,3 g/mol) et DSPC-d70 (C44H18NO870, 860,1 g/mol)35,38, de sorte que mDMPC-d54 = 133,0 mg et mDSPC-d70 = 67,0 mg. Ce schéma de contraste(Figure 2A,panneau de droite) amplifie le signal de diffusion des groupes de tête lipidiques (NSLD ~ 4,5 × 10-6 Å -2) en faisant correspondre le groupe de queue (NSLD ~ 6,4 × 10-6 Å -2) au tampon deutéré permettant la détection des fluctuations de l’épaisseur de la membrane.
- Pour les études de fluctuation d’épaisseur de certains compartiments membranaires, par exemple la matrice riche en DMPC, utilisez la même stratégie décrite à l’étape 1.2 en remplaçant les lipides DMPC protiés par leurs analogues deutérés par la queue, c’est-à-dire DMPC-d54, de sorte que les domaines riches en DSPC correspondent au contraste avec le tampon deutéré et que le signal de diffusion primaire provient de la région du groupe de tête de la matrice riche en DMPC deutérée par la queue.
 133,4 mg, où XDMPC et XDSPC sont les fractions molaires de DMPC et DSPC, ici réglés à 0,7 et 0,3, respectivement, et MwDMPC et MwDSPC sont les poids molaires donnés par 677,9 g/mol et 790,1 g/mol, respectivement. De même, mDSPC = 66,6 mg. Ce schéma de deutération augmente le contraste de diffusion entre la membrane (NSLD ~ 0 × 10-6 Å -2) et le tampon deutéré (NSLD ~ 6,4 × 10-6 Å -2) et amplifie le signal des ondulations membranaires (voir le panneau de gauche de la figure 2A).
133,4 mg, où XDMPC et XDSPC sont les fractions molaires de DMPC et DSPC, ici réglés à 0,7 et 0,3, respectivement, et MwDMPC et MwDSPC sont les poids molaires donnés par 677,9 g/mol et 790,1 g/mol, respectivement. De même, mDSPC = 66,6 mg. Ce schéma de deutération augmente le contraste de diffusion entre la membrane (NSLD ~ 0 × 10-6 Å -2) et le tampon deutéré (NSLD ~ 6,4 × 10-6 Å -2) et amplifie le signal des ondulations membranaires (voir le panneau de gauche de la figure 2A).2. Préparation de la suspension lipidique pour l’extrusion
- Calculer la masse de chaque constituant de l’échantillon, en fonction de la composition de l’échantillon. En règle générale, pour les échantillons à composantes moléculaires multiples, la masse d’un composant est donnée par sa masse molaire, Mwi, pondérée par sa fraction molaire, Xi, et normalisée sur tous les composants de telle sorte que: où
 Mtot est la masse totale, fixée ici à 200 mg. Voir l’exemple ci-dessus pour les bicouches lipidiques DMPC-DSPC avec différents schémas de deutération.
Mtot est la masse totale, fixée ici à 200 mg. Voir l’exemple ci-dessus pour les bicouches lipidiques DMPC-DSPC avec différents schémas de deutération. - À l’aide d’une semi-microbalance numérique, peser les masses calculées de lipides (et d’autres constituants de l’échantillon, par exemple des protéines, des nanoparticules, etc.) et les ajouter à un flacon ou à une fiole à fond rond – n’oubliez pas de peser le flacon ou la fiole au préalable. Ajouter 1 mL de solvant pour dissoudre les composants pesés en mélangeant manuellement à l’intérieur d’une hotte. Pour les échantillons de lipides purs, utilisez du chloroforme ou de l’éthanol. Pour les échantillons avec des composants non lipidiques supplémentaires (par exemple, des nanoparticules), choisissez un solvant commun qui disperse tous les composants.
- Pour les petites quantités de lipides (<10 mg), préparer une solution d’élevage et pipeter le volume requis dans le mélange.
REMARQUE: N’ajoutez pas de quantités excessives de solvant car cela ralentira considérablement l’étape de séchage du solvant décrite ci-dessous.
- Pour les petites quantités de lipides (<10 mg), préparer une solution d’élevage et pipeter le volume requis dans le mélange.
- Sécher la solution lipidique, à l’intérieur d’une hotte, en faisant couler doucement un gaz inerte (p. ex. azote, argon) dans le flacon tout en faisant tourner lentement le flacon en angle. Gardez les flacons en position inclinée pour créer une fine pellicule de lipides séchés sur les parois du flacon, ce qui permettra un séchage uniforme. Placez le flacon par intermittence dans un bain-marie à 35 °C pour éviter le refroidissement médié par évaporation, ce qui ralentira l’évaporation du solvant.
- Placez les flacons pendant la nuit dans un four à vide à ~35 °C pour éliminer complètement le solvant résiduel. Pour les lipides insaturés, purger le vide avec un gaz inerte pour minimiser l’oxydation.
- Pour assurer l’élimination complète du solvant, peser le flacon après séchage des lipides et confirmer qu’il n’y a pas d’excès de masse au-delà des quantités mesurées de matériaux. Pour ce faire, soustrayez la masse du flacon de la masse mesurée après séchage. S’il y a un excès de masse, sécher l’échantillon sous vide pendant encore 6 h. Répétez ce processus si nécessaire.
- Hydrater le film lipidique avec 4 mL de tamponD2O ouD2O pour obtenir une concentration lipidique de 50 mg/mL. Pour les lipides à haute température de transition, tels que les mélanges DMPC-DSPC, chauffer le tampon au-dessus de la température de transition (60 °C) pour assurer un mélange uniforme.
REMARQUE: Étant donné que les expériences NSE nécessitent des volumes d’échantillon relativement importants (~ 4 mL), envisagez d’hydrater l’échantillon en utilisant la moitié du tampon requis, c’est-à-dire 2 mL, afin de minimiser le nombre d’extrusions par échantillon (voir rubrique 3). Dans ce cas, ajoutez la moitié restante de la post-extrusion tampon. Notez que la capacité des seringues utilisées dans l’extrusion est limitée à 1 mL. Ainsi, l’hydratation avec 4 mL de tampon nécessiterait quatre ensembles d’extrusion. - Mélangez en vortex la solution lipidique hydratée jusqu’à ce que le film lipidique soit complètement dissous et ne soit plus visible sur les parois du flacon. À ce stade, les lipides hydratés forment des vésicules multilamellaires et des piles multilamellaire de la taille d’un micron et la suspension apparaît d’un blanc laiteux.
- Pour faciliter la rupture des piles lipidiques et réduire la multilamellité, effectuer cinq cycles de congélation/décongélation en plaçant le flacon de solution lipidique hydratée dans un congélateur de laboratoire (de préférence un congélateur de qualité -80 °C) jusqu’à ce qu’il soit complètement congelé, puis en transférant le flacon dans un bain-marie à 35 °C jusqu’à ce que la solution lipidique soit complètement décongelée. Vortex la solution décongelée jusqu’à homogénéisme. Répétez quatre fois de plus.
REMARQUE: Alternativement, un bain de glace carbonique peut être préparé pour une congélation rapide en combinant de l’acétone et de la glace sèche.
 Mtot est la masse totale, fixée ici à 200 mg. Voir l’exemple ci-dessus pour les bicouches lipidiques DMPC-DSPC avec différents schémas de deutération.
Mtot est la masse totale, fixée ici à 200 mg. Voir l’exemple ci-dessus pour les bicouches lipidiques DMPC-DSPC avec différents schémas de deutération.3. Extrusion de la solution lipidique hydratée
- Assemblez la configuration de l’extrudeuse à l’aide d’une membrane en polycarbonate entre deux supports de membrane et en ajoutant deux filtres en papier de chaque côté pour fournir un support supplémentaire. Utilisez une membrane en polycarbonate avec une taille de pore qui correspond à la taille liposomale cible (les tailles de pores courantes pour les expériences NSE sont de 50 nm et 100 nm – généralement, les liposomes de 100 nm de diamètre permettent des fluctuations membranaires moins contraintes, mais des liposomes plus petits de 50 nm pourraient être utilisés pour les études de courbure). Assurez-vous que la membrane en polycarbonate est complètement étirée avant de terminer l’assemblage et de serrer le boîtier externe de l’extrudeuse.
- Hydrater la membrane en polycarbonate en passant environ 0,3 mL de tampon D2O ou D2O à travers l’assemblage de la membrane à l’aide de seringues en verre hermétiques. Utilisez le même tampon que celui utilisé dans la préparation des échantillons. Laissez-le pendant au moins 10 minutes, puis aspirez complètement le tampon avant d’introduire l’échantillon.
- Remplissez une seringue étanche aux gaz de 1 mL avec la solution lipidique préparée et insérez-la dans une extrémité de l’appareil d’extrusion. Ensuite, insérez une seringue vide à l’extrémité opposée. Une fois les seringues connectées à l’ensemble de l’extrudeuse, placez-le dans le bloc de l’extrudeuse.
- Si des températures élevées sont nécessaires pour l’extrusion, comme dans le cas de lipides saturés avec des températures de transition élevées (par exemple, DSPC, Tm = 55 °C), préchauffez le bloc chauffant de l’extrudeuse au-dessus de la température de transition lipidique (par exemple, 60 °C), en plaçant le bloc chauffant sur une plaque chauffante ou en utilisant un bain de circulation comme indiqué à la figure 3A.
REMARQUE: Cette étape est cruciale pour assurer un mélange homogène des lipides et éviter d’exercer une pression extrême lors de l’extrusion, ce qui pourrait rompre la membrane en polycarbonate. Pour les échantillons lipidiques à basse température de transition (<25 °C), effectuez l’extrusion à température ambiante. - Pour extruder la solution lipidique, fixez l’ensemble de l’extrudeuse à une pompe à seringue programmable avec un cadre en aluminium/acier, comme illustré à la figure 3A. Pour les extrusions à température contrôlée, ajoutez une base d’extrudeuse sur mesure avec un canal de fluide et fixez-la à un bain-marie circulant.
- Programmez la pompe à seringue pour effectuer 15 à 20 cycles d’extrusion en suivant le manuel du fabricant. Une fois extrudée, la couleur de la solution lipidique passe du blanc laiteux au bleu opale transparent(Figure 3B,C),indiquant une taille liposomale finale inférieure à la longueur d’onde de la lumière visible, comme prévu. Pour le type de pompe à seringue illustré à la figure 3A,suivez les étapes ci-dessous.
- Commencez par ajuster les paramètres de la pompe. Maintenez le bouton Taux enfoncé et entrez le taux d’extrusion (50,99 mL/h), puis appuyez sur le bouton Diamètre et entrez le diamètre de la seringue (4,606 mm). Utilisez les flèches vers le haut sous chaque chiffre à l’écran pour modifier la valeur de ce chiffre.
- Placez l’ensemble d’extrudeuses avec la seringue d’échantillonnage vers la droite (voir Figure 3A). Appuyez sur le bouton Retirer jusqu’à ce que le voyant de retrait s’allume. Appuyez sur Démarrer et attendez que l’échantillon soit distribué dans la seringue gauche (vide).
- Appuyez sur le bouton Arrêter juste avant que la seringue d’échantillon (droite) ne soit complètement vide. Enregistrez le volume distribué et utilisez-le pour programmer le cycle d’extrusion. Maintenez le bouton Taux enfoncé jusqu’à ce que la phase 1 (PH:01) apparaisse à l’écran. Appuyez sur le bouton Volume pour entrer le volume distribué enregistré précédemment. Dans cette phase, assurez-vous que la lumière Withdraw est éteinte – cela distribue l’échantillon dans la bonne direction.
- Appuyez à nouveau sur le bouton Taux et utilisez la flèche la plus à droite vers le haut pour accéder à la phase 2 (PH:02). Appuyez sur Volume pour entrer la même valeur que le volume distribué enregistré précédemment. Dans cette phase, appuyez sur le bouton Retirer jusqu’à ce que le voyant Retirer soit allumé – cela distribue l’échantillon à gauche.
- Pour répéter ce cycle, appuyez à nouveau sur le bouton Taux et utilisez la flèche vers le haut la plus à droite pour accéder à la phase 3 (PH:03). Appuyez sur le bouton Volume jusqu’à ce que LP:SE apparaisse à l’écran et réglez-le sur 20. Il s’agit du nombre de boucles ou de répétitions que la pompe effectuera. Enfin, appuyez sur le bouton Taux, accédez à la phase 4 (PH: 04) et appuyez sur le bouton Volume pour accéder à la fonction Arrêter. La pompe est maintenant configurée pour l’extrusion automatisée.
- Appuyez sur Démarrer pour démarrer le cycle d’extrusion.
- Videz la seringue contenant la suspension lipidique extrudée dans un flacon propre et préparez-la pour le stockage ou les mesures. Pour les échantillons lipidiques à température de fusion élevée, conservez l’échantillon au-dessus de la transition de phase du fluide jusqu’à mesure qu’il soit mesuré. Sinon, conservez les échantillons à température ambiante.
- Ne congelez pas les échantillons extrudés, car la congélation provoquera l’éclatement des vésicules (la suspension redevisera blanc laiteux).
4. Mesures NSE pour le(s) échantillon(s) et réduction des données collectées
- Avant l’expérience NSE, caractériser l’échantillon liposomal extrudé de l’étape 3.7 en utilisant les méthodes disponibles pour assurer une qualité d’échantillon adéquate. Une liste des méthodes de charcatérisation potentielles qui peuvent être utilisées pour évaluer la qualité des suspensions liposomales pour les expériences NSE, par exemple, la distribution de taille, la multilamellaire, la structure de la membrane latérale, est incluse dans la section de discussion.
- Déterminez la plage Q et les paramètres d’instrument correspondants requis pour l’expérience. Pour les mesures de rigidité en flexion des bicouches lipidiques, utilisez une plage Q de ~(0,04 - 0,2) Å-1. Pour les études des fluctuations d’épaisseur de membrane, utilisez une plage Q de ~(0,04 - 0,2) Å-1 correspondant à l’épaisseur de membrane35,66,67.
REMARQUE: Discutez de la configuration expérimentale avec le scientifique de l’instrument avant le début de l’expérience. Comme mentionné précédemment, la caractérisation SANS de l’échantillon est nécessaire, en particulier si l’information préalable du signal de diffusion n’est pas disponible, comme dans les membranes deutérées sélectivement. Alternativement, exécutez des mesures statiques (également connues sous le nom de diffraction) sur une plage Q limitée sur l’instrument NSE, avec la mise en garde que ces mesures prennent beaucoup plus de temps que SANS. - À l’aide d’une seringue ou d’une pipette de transfert, chargez la ou les suspensions liposomales extrudées dans les cellules d’échantillon désignées disponibles aux lignes de faisceau NSE. Notez que les cellules d’échantillon NSE standard sont disponibles en épaisseurs de 1, 2, 3 et 4 mm. Choisissez l’épaisseur de la cellule de manière à optimiser le signal de diffusion tout en maintenant le signal de fond incohérent à une intensité raisonnable.
REMARQUE: En règle générale, utilisez des cellules d’échantillon avec une longueur de trajet de 1 ou 2 mm pour les liposomes protisés dans un tampon deutéré - des cellules plus épaisses pourraient entraîner de multiples effets de diffusion difficiles à corriger. Pour les liposomes présentant des niveaux plus élevés de deutérations (p. ex., liposomes appariés au contraste de la queue ou liposomes asymétriques avec des feuillets protifés uniques), envisagez d’utiliser une cellule d’échantillon plus épaisse (p. ex., longueur de chemin de 3 ou 4 mm) pour améliorer les statistiques de comptage si l’échantillon est disponible en plus grandes quantités – parfois, cela peut être prohibitif. - Préparez une cellule d’échantillon identique pour le tampon. Utilisez le même tampon que dans la suspension liposomale. Des mesures sur le tampon sont nécessaires pour la normalisation de l’intensité et les corrections de fond (BKG).
- Placez la ou les cellules d’échantillon dans le porte-échantillon du spectromètre NSE, programmez les séries de mesure et collectez les données d’écho. Consultez le scientifique de l’instrument au sujet de la programmation des mesures si vous utilisez un nouvel utilisateur de NSE.
- Effectuez deux ensembles supplémentaires de mesures nécessaires à la réduction des données : les mesures de résolution (R) et de transmission (T).
- Effectuer une mesure de résolution (R) sur une référence de diffusion élastique (par exemple, le carbone) - à exécuter dans les mêmes paramètres; c’est-à-dire les mêmes vecteurs d’onde et les mêmes temps de Fourier que les mesures d’échantillon et de tampon.
- Effectuer des mesures de transmission (T) sur l’échantillon et le tampon pour calculer l’intensité du faisceau de neutrons transmis (voir l’étape 4.9. ci-dessous). La transmission est calculée comme le rapport des nombres de neutrons de l’échantillon ou du tampon divisé par les comptes de neutrons pour un faisceau ouvert (c.-à-d. avec une position d’échantillon vide).
- Utilisez le logiciel de réduction de données dédié pour le spectromètre NSE sur lequel les mesures sont effectuées afin de réduire les données collectées.
REMARQUE: Différents spectromètres peuvent utiliser différents logiciels ou interfaces utilisateur. Vous trouverez ci-dessous un exemple de réduction des données NSE à l’aide de l’environnement d’analyse et de visualisation des données (DAVE)68 logiciel spécialement écrit pour le spectromètre NSE du NIST Center for Neutron Research.- Ouvrez le logiciel DAVE et sélectionnez Réduire les données NSE dans le menu de réduction des données. Plusieurs fenêtres pop-up apparaîtront.
- Téléchargez les fichiers de données sur différentes valeurs Q à l’aide du fichier Ouvrir .echo dans le menu Fichier. Ces fichiers correspondent aux fichiers de données brutes avec les signaux d’écho de spin et ont l’extension .echo dans le nom du fichier. Une fois le téléchargement du fichier terminé, les fichiers s’afficheront sous les ensembles de données disponibles.
- Faites un clic droit sur le fichier sélectionné et étiquetez-le en fonction de la mesure à laquelle il correspond; c’est-à-dire Échantillon, Cellule (pour cellule vide ou tampon) ou Résolution.
- Regroupez les pixles du détecteur en 2 x 2 pour améliorer le rapport signal/bruit à l’aide de l’onglet Ensemble de données. Appliquez le même binning à tous les fichiers ; c’est-à-dire Résolution, Cellule et Échantillon.
- Inspectez les données sur tous les groupes de pixels et masquez ceux dont les signaux sont faibles (voir Figure 4B)en appuyant sur la touche m du clavier. Appuyez sur Entrée pour accéder à une fenêtre contextuelle afin d’appliquer le même masque à toutes les fois de Fourier ou aux heures de Fourier suivantes. Cela peut également être appliqué à des pixels individuels à tout moment pendant la réduction des données. Les pixels masqués deviennent verts.
- S’assurer que les données recueillies se présentent sous la forme d’un signal d’écho, c’est-à-dire d’une fonction cosinus en termes de courant de phase, sur chaque pixel du détecteur (voir la figure 4A).
NOTE: Le courant de phase est proportionnel à l’angle de précession du spin du neutron; par conséquent, il est courant de représenter le courant de phase sous la forme d’un angle de phase, comme le montre la figure 4A. Pour les mesures sur des sources pulsées, des calculs de temps de vol supplémentaires sont appliqués aux données pour obtenir les signaux d’écho en fonction de la longueur d’onde des neutrons incidents dans une impulsion neutronique. - Commencez par ajuster le fichier de résolution. Sélectionnez un fichier de résolution dans la liste des fichiers téléchargés et cliquez avec le bouton droit sur le fichier. Sélectionnez Ajuster les opérations : Ajuster les échos (résolution) dans le menu contextuel.
- Assurez-vous que les ajustements des signaux d’écho produisent un certain nombre de paramètres d’ajustement, y compris le paramètre A, requis à l’étape 4.8. Les ajustements sont automatiquement effectués à l’aide de l’expression suivante.

Ici, ζ est la période du signal d’écho (c’est-à-dire la fonction cosinus de la figure 4A),σ est la largeur de l’enveloppe gaussienne déterminée par la longueur d’onde moyenne et la propagation de longueur d’onde du faisceau de neutrons incident, Φc est le courant de phase et Φ0 est le point d’écho qui dépend du chemin de champ expérimenté par les neutrons50. Les informations physiques sur l’échantillon sont codées dans l’amplitude Ade la fonction cosinus dans l’équation (1).
REMARQUE: La largeur de l’enveloppe gaussienne est basée sur des valeurs prédéterminées par le scientifique de l’instrument et ne doit pas être modifiée. Les autres paramètres sont des variables qui sont ajustées au signal d’écho spécifique sur chaque pixel. - Inspectez les résultats de l’ajustement en cliquant sur chaque pixel pour afficher les paramètres d’ajustement résultants, la qualité de l’ajustement et l’écart carré moyen de l’ajustement. Pour inspecter l’erreur associée à chaque paramètre de raccord sur l’ensemble du détecteur, sélectionnez Options d’image, puis sélectionnez le paramètre d’ajustement qui vous intéresse. Cela générera une carte avec la valeur du paramètre d’ajustement sur chaque pixel. Faites un clic droit sur l’image du détecteur. Une fenêtre contextuelle apparaîtra montrant une carte de barre d’erreur du paramètre d’ajustement sélectionné.
- Si l’ajustement sur un pixel spécifique n’est pas satisfaisant (par exemple, ajuster des paramètres avec de grandes barres d’erreur), réajustez le signal sur ce pixel spécifique. Sélectionnez ce pixel, appuyez sur l’onglet Raccord, puis appuyezsur Ajuster le pixel . Entrez de nouveaux paramètres de départ pour la phase (Φ0) et la période (ζ) dans l’onglet Raccord pour obtenir un ajustement plus satisfaisant.
REMARQUE: Il est utile de tracer la phase ajustée en fonction du temps de Fourier. Pour ce faire, accédez à la fenêtre principale du tracé et sélectionnez Ajuster la phase v. Temps de Fourier. Cette intrigue doit être lisse et continue. Inspectez les discontinuités dans ce tracé et réajustez les pixels auxquels elles correspondent.
- Réduisez le fichier Sample ou Cell en sélectionnant le fichier correspondant dans la liste des fichiers téléchargés et étiquetés.
- Inspectez tous les pixels et masquez ceux qui ont de mauvaises statistiques comme décrit à l’étape 4.7.5.
- Cliquez avec le bouton droit sur le fichier et sélectionnez Ajuster les opérations : Phases d’importation (exemple, cellule). Cela importe les phases et le masque appliqué à partir du fichier de résolution.
- Ajustez les signaux d’écho en utilisant la même procédure décrite précédemment pour le fichier de résolution (étapes 4.7.8-4.7.10). Lors de l’ajustement des fichiers Sample et Cell, ne modifiez pas les valeurs de la période et du point de phase d’écho importés à partir des ajustements de résolution. Ces paramètres dépendent des paramètres instrumentaux et ne doivent pas varier d’un échantillon à l’autre.
- Avant de procéder à la réduction des données, entrez le centre de faisceau pour tous les fichiers de données. Sélectionnez le fichier de données, accédez à l’onglet Général et entrez les valeurs centrales des poutres X et Y. Ces valeurs sont enregistrées au cours de l’expérience.
- Une fois les ajustements aux fichiers Échantillon, Cellule et Résolution terminés, calculez la fonction de diffusion intermédiaire normalisée à utiliser ultérieurement dans l’analyse et l’interprétation des données. Pour ce faire, faites un clic droit sur le fichier Exemple à réduire dans la liste des fichiers ajustés, puis sélectionnez Calculer I(Q) dans le menu contextuel. Une fenêtre apparaîtra avec des choix d’entrée pour les fichiers Résolution et Cellule (c’est-à-dire mémoire tampon) et le nombre d’arcs Q (voir l’étape 4.9). Après avoir entré toutes les informations requises, appuyez sur le bouton OK. Les résultats apparaîtront dans une nouvelle fenêtre.
REMARQUE: La réduction des données est effectuée selon l’équation suivante pour obtenir la fonction de diffusion intermédiaire normalisée69.
où t est le temps de Fourier, N vers le haut et N vers le bas sont les nombres de neutrons dans les configurations sans spin-flip et spin-flip (mesurées avec les π/2-flippers désactivés et les π-flipper éteints et activés, respectivement), et les exposants, BKG et R, correspondent respectivement aux mesures d’arrière-plan et de résolution, telles que définies aux étapes 4.4 et 4.6. Notez que la polarisation du faisceau, donc les changements dans l’état de spin dus à l’échange d’énergie entre le neutron et l’échantillon est détectée comme une baisse de la polarisation (de l’unité).
faisceau, donc les changements dans l’état de spin dus à l’échange d’énergie entre le neutron et l’échantillon est détectée comme une baisse de la polarisation (de l’unité).
- Enfin, regroupez les pixels du détecteur en arcs Q-comme le montre la figure 4B pour obtenir la Q-dépendancede la fonction de diffusion intermédiaire normalisée, S(Q,t)/ S(Q,0). C’est ce qu’on appelle techniquement le regroupement de données et cela doit être fait judicieusement, c’est-à-dire en tenant compte des statistiques de comptage de l’échantillon et de l’écart-type attendu des données sur les pixels groupés.
- Pour les échantillons à forte diffusion, divisez le détecteur en plus d’arcs Q tout en maintenant des barres d’erreur raisonnables sur la fonction de diffusion intermédiaire résultante, S(Q,t) / S(Q, 0 ). Cela donne plus de points de données Q et est important pour la procédure d’analyse des données décrite ci-dessous. Sachez que pour les échantillons à faible diffusion, un binning excessif entraîne de mauvais signaux de désintégration, c’est-à-dire de grandes barres d’erreur sur S(Q,t) / S(Q, 0 ), ce qui pourrait entraîner de grandes incertitudes.


 faisceau, donc les changements dans l’état de spin dus à l’échange d’énergie entre le neutron et l’échantillon est détectée comme une baisse de la polarisation (de l’unité).
faisceau, donc les changements dans l’état de spin dus à l’échange d’énergie entre le neutron et l’échantillon est détectée comme une baisse de la polarisation (de l’unité).5. Analyse et interprétation des données
- Ajuster les fonctions de diffusion intermédiaire normalisées, S(Q,t) / S(Q, 0 ), obtenues à partir de la réduction des données ci-dessus à une fonction exponentielle étirée avec un exposant d’étirement de 2/370.

REMARQUE : Un exemple de ces ajustements est fourni à la figure 5B. Les ajustements de S(Q,t) / S(Q, 0 ) à l’équation (3) donnent les taux de relaxation dépendants de Q Γ (Q). - Tracer Γ(Q) en fonction de Q et s’adapter à un modèle approprié pour extraire les paramètres de membrane pertinents.

Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Les études NSE accédant aux fluctuations de flexion sont généralement réalisées sur une plage Q de ~ (0,04 - 0,2) Å-1. Cette gamme Q correspond à des échelles de longueur intermédiaires entre l’épaisseur de la membrane et le rayon liposomal, où la dynamique de flexion domine. La mesure sur une plage Q étendue peut donner accès à des modes dynamiques supplémentaires, y compris la diffusion liposomale et la dynamique intramembranaire. Pour plus de détails sur le cross-over dans la dynamique membranaire accessible par NSE, consultez ces publications pertinentes25,71. Il est important de souligner que les signaux NSE sont proportionnels à:  , où Icoh et Iinc sont, respectivement, l’intensité de diffusion cohérente et incohérente de l’échantillon. Par conséquent, il est conseillé de préparer des échantillons liposomaux NSE dans des tampons deutérés (c’est-à-dire des tampons préparés avec D2O au lieu de H2O) afin de minimiser le signal de diffusion incohérent, principalement contribué par la teneur en hydrogène de l’échantillon. Cependant, dans certains cas, des schémas de deutération intermédiaires (c’est-à-dire utilisant des mélanges deD2O et H2O) peuvent être nécessaires pour obtenir des conditions de contraste optimales. En règle générale, les mesures NSE des fluctuations de flexion de la membrane sont effectuées sur des liposomes entièrement protiés dans un tampon deutéré, appelés liposomes entièrement contrastés dans la figure 5. Ce schéma de deutération entraîne une grande différence NSLD entre le noyau de la membrane (~0 × 10-6 Å-2) et son environnement fluide deutéré (~6,4 × 10-6 Å -2), ce qui améliore considérablement le signal de diffusion des membranes liposomales et améliore les statistiques de mesure de la dynamique de flexion. Ce schéma de contraste(figure 2A panneau de gauche) est fréquemment utilisé dans les études de rigidité de flexion des membranes lipidiques avec des composants lipidiques simples38,72 et multiples39,66 et dans les études de ramollissement / raidissement de la membrane par des inclusions biologiques (par exemple, cholestérol, molécules médicamenteuses, peptides / protéines)36,37,73,74,75et des additifs synthétiques (par exemple, nanoparticules)76,77.
, où Icoh et Iinc sont, respectivement, l’intensité de diffusion cohérente et incohérente de l’échantillon. Par conséquent, il est conseillé de préparer des échantillons liposomaux NSE dans des tampons deutérés (c’est-à-dire des tampons préparés avec D2O au lieu de H2O) afin de minimiser le signal de diffusion incohérent, principalement contribué par la teneur en hydrogène de l’échantillon. Cependant, dans certains cas, des schémas de deutération intermédiaires (c’est-à-dire utilisant des mélanges deD2O et H2O) peuvent être nécessaires pour obtenir des conditions de contraste optimales. En règle générale, les mesures NSE des fluctuations de flexion de la membrane sont effectuées sur des liposomes entièrement protiés dans un tampon deutéré, appelés liposomes entièrement contrastés dans la figure 5. Ce schéma de deutération entraîne une grande différence NSLD entre le noyau de la membrane (~0 × 10-6 Å-2) et son environnement fluide deutéré (~6,4 × 10-6 Å -2), ce qui améliore considérablement le signal de diffusion des membranes liposomales et améliore les statistiques de mesure de la dynamique de flexion. Ce schéma de contraste(figure 2A panneau de gauche) est fréquemment utilisé dans les études de rigidité de flexion des membranes lipidiques avec des composants lipidiques simples38,72 et multiples39,66 et dans les études de ramollissement / raidissement de la membrane par des inclusions biologiques (par exemple, cholestérol, molécules médicamenteuses, peptides / protéines)36,37,73,74,75et des additifs synthétiques (par exemple, nanoparticules)76,77.
Les mesures des fluctuations de flexion entraînent des taux de relaxation qui suivent une dépendance Q3, comme prédit par Zilman et Granek pour les feuilles minces élastiques ondulantes thermiquement70. Une forme affinée de cette Q-dépendanceest obtenue à partir de corrections théoriques de Watson et Brown78,qui prennent en compte les effets du frottement intermonouche proposé par Seifert et Langer79. En définissant en outre le plan neutre comme faisant partie de l’interface entre les groupes de têtes hydrophiles et les queues hydrophobes de la membrane, les taux de relaxation en flexion peuvent alors être ajustés à l’expression suivante38.

où ηbuff est la viscosité tampon, kBT est l’énergie thermique, κ et est la rigidité de flexion de la membrane mesurée (ou de la partie contrastée de la membrane dans les systèmes deutérés sélectivement). Ce type de mesure permet le calcul direct des propriétés élastiques de la membrane sous la forme du module de rigidité de flexion. Notez que κ est extrait de la pente de l’ajustement linéaire de Γ vs Q3, comme le montre la figure 5C.
D’autre part, les mesures NSE des fluctuations d’épaisseur de membranemontrent des écarts par rapport à la dépendance Q3dans Γ( Q ) autour de valeurs Q qui correspondent à l’épaisseur de la membrane (voir figure 2 dans la réf.66). Pour isoler le signal de fluctuation d’épaisseur, on peut diviser Γ(Q) par Q3, comme le montre la figure 5D. Les données résultantes montrent que la dynamique excédentaire due aux fluctuations d’épaisseur suit une fonction lorentzienne dans Q, comme récemment corroboré dans les simulations de dynamique moléculaire à grains grossiers (MD)67. Pour s’adapter à la dynamique excessive observée, Nagao et al.38 ont développé une expression basée sur le cadre théorique des fluctuations membranaires par Bingham et al.80 comme suit.

Dans cette expression, Q0 est la valeur Qde crête correspondant à l’épaisseur de la membrane (qui peut être obtenue indépendamment à partir de mesures SANS), μ est la viscosité de la membrane dans le plan, AL est la surface par lipide (mesurée avec SANS/SAXS), et KA est le module de compressibilité de l’aire. En supposant que KA puisse être calculé à partir de κ à l’aide du modèle de brosse polymère, cette expression se réduit à un paramètre d’ajustement, à savoir la viscosité de la membrane μ, présentant une nouvelle approche pour mesurer la viscosité de la membrane sans avoir besoin de marquage de fluorescence ou d’attache/suivi des particules13. La prémisse est que selon les modèles de déformation des feuilles minces élastiques81, κ et KA sont interdépendantes telles que: ,  où tm est l’épaisseur mécanique (ou déformable) de la membrane et β est une constante qui décrit le couplage interleaflet. L’hypothèse est que β = 12 pour les folioles entièrement couplées, β = 48 pour les folioles complètement découplées et β = 24 pour les folioles à couplage intermédiaire. Ce dernier est appelé le modèle de brosse polymère81 et il a été démontré qu’il s’applique dans les membranes lipidiques fluides monocomposantes et binaires39. Cependant, cela doit être abordé avec prudence. Par exemple, des simulations récentes de Doktorova et al. 82 a montré que pour que le modèle de brosse en polymère maintienne dans des membranes lipidiques insaturées contenant du cholestérol, une expression modifiée de l’épaisseur mécanique de la membrane doit être utilisée. Idéalement, si une mesure indépendante de KA est possible, par exemple en utilisant l’aspiration par micropipette83, la combinaison des résultats KA avec des mesures de rigidité de flexion NSE présenterait une occasion unique d’étudier le couplage entre les feuilles dans les membranes modèles et biologiques – une question de longue date en biophysique membranaire et en biologie structurale. Une fois les valeurs de KA validées, elles peuvent être utilisées dans l’équation 5 pour obtenir la viscosité mésoscopique de la membrane.
où tm est l’épaisseur mécanique (ou déformable) de la membrane et β est une constante qui décrit le couplage interleaflet. L’hypothèse est que β = 12 pour les folioles entièrement couplées, β = 48 pour les folioles complètement découplées et β = 24 pour les folioles à couplage intermédiaire. Ce dernier est appelé le modèle de brosse polymère81 et il a été démontré qu’il s’applique dans les membranes lipidiques fluides monocomposantes et binaires39. Cependant, cela doit être abordé avec prudence. Par exemple, des simulations récentes de Doktorova et al. 82 a montré que pour que le modèle de brosse en polymère maintienne dans des membranes lipidiques insaturées contenant du cholestérol, une expression modifiée de l’épaisseur mécanique de la membrane doit être utilisée. Idéalement, si une mesure indépendante de KA est possible, par exemple en utilisant l’aspiration par micropipette83, la combinaison des résultats KA avec des mesures de rigidité de flexion NSE présenterait une occasion unique d’étudier le couplage entre les feuilles dans les membranes modèles et biologiques – une question de longue date en biophysique membranaire et en biologie structurale. Une fois les valeurs de KA validées, elles peuvent être utilisées dans l’équation 5 pour obtenir la viscosité mésoscopique de la membrane.

Figure 1: Conception del’instrument NSE et chevauchement synergique avec les échelles longueur/temps de la dynamique membranaire mésoscopique. (A) Schéma des différents éléments magnétiques d’un instrument NSE, utilisé pour manipuler le spin des neutrons traversant l’instrument de gauche à droite. Le neutron mis en évidence indique un changement d’orientation de spin (ou une perte de polarisation) dû à l’échange d’énergie entre le neutron et l’échantillon, tandis que le neutron transparent représente l’écho de spin, c’est-à-dire aucun changement dans le spin du neutron dû à un échange d’énergie nul. La flèche grise indique la possibilité de faire pivoter le deuxième bras du spectromètre pour accéder à des angles de diffusion plus grands. (B) Représentation picturale de la dynamique hiérarchique dans les membranes lipidiques, montrant divers modes dynamiques qui s’étendent sur plusieurs échelles de longueur et de temps. La zone ombrée représente les échelles de longueur et de temps auxquelles accède NSE, qui chevauchent les méso-échelles des fluctuations membranaires collectives, à savoir les fluctuations de flexion et d’épaisseur. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 2: Exemples de schémas de deutération possibles dans les expériences NSE sur membranes lipidiques. (A) Gauche: Membranes entièrement contrastées, par exemple membranes protées dans un tampon deutéré, montrant le profil NSLD le long de la normale à la surface de la membrane. La différence dans la NSLD entre la région de la queue (~0 × 10-2 Å-2) et la région du groupe de tête (~4,5 × 10-6 Å-2) de la membrane est due à l’hydratation du groupe de tête avec tampon deutéré. Droite : Membranes appariées au contraste de la queue de telle sorte que la région de la queue d’hydrocarbure de la membrane a la même LNDS que le tampon, comme indiqué dans le profil NSLD correspondant le long de la membrane normale. (B) Membranes formant un domaine avec deux schémas de contraste neutronique où les domaines (au centre) ou la matrice (à gauche) sont assortis au tampon, permettant des études sélectives de la matrice ou de la dynamique du domaine, respectivement. Cette figure a été modifiée à partir de Nickels et al., JACS 201541. (C) Membranes asymétriques préparées par échange de cyclodextrine entre des vésicules lipidiques protées et deutérées, entraînant la deutération d’une feuillet membranaire tout en maintenant l’autre feuillet protiée. Cela permet d’études de la dynamique de flexion de la feuillet protiée et fournit des informations sur le couplage mécanique entre les folioles opposées dans les membranes asymétriques. Cette figure a été modifiée à partir de Rickeard et al., Nanoscale 202040. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 3: Illustration de la configuration pour l’extrusion automatisée de liposomes. (A) Extrudeuse automatisée sur mesure utilisant une pompe à seringue, un mini ensemble d’extrudeuses et un cadre en aluminium / acier pour permettre des extrusions cycliques. (B) et (C) montrent la différence d’aspect visuel des suspensions lipidiques avant (blanc laiteux) et après (bleu opale transparent) extrusion. Cela est dû à la formation initiale de piles lipidiques de la taille d’un micron ou de vésicules géantes qui sont de l’ordre ou supérieures à la longueur d’onde de la lumière visible. Après extrusion, la suspension comprendra des vésicules nanoscopiques (~100 nm), qui sont plus petites que la longueur d’onde de la lumière visible, donnant une suspension transparente. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 4: Données représentatives d’expériences NSE sur suspensions liposomiques. (A) Exemple d’un signal d’écho sur un seul pixel de détecteur (pixel marqué dans le panneau B), montrant les ajustements du signal d’écho à l’aide de l’équation (1), avec une illustration des différents paramètres requis dans l’ajustement de l’écho. Notez que le signal d’écho est tracé en fonction de l’angle de phase au lieu du courant de phase comme indiqué à l’étape 4.7 du protocole. (B) Image du détecteur NSE montrant la variation du nombre de neutrons par pixel. L’image montre également des pixels de détecteur éliminés (vert) en raison de mauvais signaux d’écho. Le binning des pixels du détecteur dans les arcs Q (également connus sous le nom d’anneaux de Debye-Scherrer) donne la Q-dépendance de la fonction de diffusion intermédiaire, nécessaire à l’analyse et à l’interprétation des données NSE. Cette figure a été modifiée à partir de Ashkar, J. Appl. Phys. 202050. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 5: Résultats représentatifs d’expériences NSE sur suspensions liposomales avec différents schémas de deutération. (A) Géométrie de diffusion d’un neutron interagissant avec un liposome, montrant l’angle de diffusion, 2θ, et le transfert du vecteur d’onde,  . (B) Les fonctions de diffusion intermédiaires, S(Q,t) / S(Q,0), présentent des désintégrations en fonction du temps de Fourier. L’ajustement des désintégrations mesurées à une fonction exponentielle étirée donnée par l’équation 3 donne les taux de relaxation, Γ(Q). (C) Pour les liposomes entièrement protiés dans un tampon deutéré, Γ(Q) suit une dépendance Q3, typique de la dynamique de flexion. L’ajustement linéaire des données obtenues à un modèle de Zilman-Granek donne le module de rigidité de flexion de la membrane. (D) Pour les liposomes deutérés de la queue, une dynamique excessive est observée en plus des fluctuations de flexion et sont plus prononcées à des valeurs Q qui correspondent à l’épaisseur de la membrane. L’ajustement de l’excès de dynamique à une fonction lorentzienne (équation 5) permet d’extraire la viscosité de la membrane. Des ensembles de données ont été recueillis sur le spectromètre NSE du NIST. Veuillez cliquer ici pour voir une version agrandie de cette figure.
. (B) Les fonctions de diffusion intermédiaires, S(Q,t) / S(Q,0), présentent des désintégrations en fonction du temps de Fourier. L’ajustement des désintégrations mesurées à une fonction exponentielle étirée donnée par l’équation 3 donne les taux de relaxation, Γ(Q). (C) Pour les liposomes entièrement protiés dans un tampon deutéré, Γ(Q) suit une dépendance Q3, typique de la dynamique de flexion. L’ajustement linéaire des données obtenues à un modèle de Zilman-Granek donne le module de rigidité de flexion de la membrane. (D) Pour les liposomes deutérés de la queue, une dynamique excessive est observée en plus des fluctuations de flexion et sont plus prononcées à des valeurs Q qui correspondent à l’épaisseur de la membrane. L’ajustement de l’excès de dynamique à une fonction lorentzienne (équation 5) permet d’extraire la viscosité de la membrane. Des ensembles de données ont été recueillis sur le spectromètre NSE du NIST. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
NSE est une technique puissante et unique dans la mesure de la dynamique mésoscopique des membranes lipidiques dans diverses conditions. L’utilisation efficace de la NSE dépend de la qualité de l’échantillon, du contraste des neutrons et de la gamme de dynamiques accessibles qui peuvent être sondées pour un échantillon donné. Ainsi, plusieurs étapes critiques sont nécessaires pour mener à bien des expériences NSE et collecter des données de haute qualité. Une étape clé pour assurer l’utilisation efficace du temps de faisceau de neutrons au cours d’une expérience NSE consiste à caractériser les suspensions liposomales avec des méthodes de laboratoire avant l’expérience NSE. Par exemple, la distribution granulométrique (ou constante de diffusion) des liposomes extrudés peut être déterminée par diffusion dynamique de la lumière (DLS), facilement disponible dans des laboratoires individuels ou dans des installations partagées84. La cryo-microscopie électronique est une autre méthode de caractérisation récemment validée sur des échantillons liposomaux, où des images de microscopie à haute résolution sur des sections cryomicrotomées de suspension liposomale peuvent être utilisées efficacement pour examiner l’unilamellité liposomale65,la formation de domaine85,86,ou l’incorporation d’additifs tels que les nanoparticules76 et les protéines87. Alternativement, la diffusion des rayons X à petit angle (SAXS) peut être utilisée pour caractériser la structure de la membrane88, évaluer la multilamellité liposomale65, ou évaluer les effets des additifs sur les propriétés structurelles de la membrane89. Outre ces techniques en laboratoire, il est fortement conseillé que les mesures NSE sur des échantillons liposomaux soient associées à des études structurelles utilisant la diffusion neutronique à petit angle (SANS)54,90. SANS est un excellent complément à NSE, non seulement pour acquérir des informations sur la membrane structurelle, mais aussi pour examiner l’intensité du signal de diffusion des neutrons de l’échantillon, confirmer le schéma de contraste et faire un choix éclairé sur la plage Q sur laquelle les mesures NSE doivent être effectuées. Par conséquent, il est recommandé que les utilisateurs de NSE demandent un temps de faisceau SANS lorsqu’ils postulent pour des expériences NSE.
Cependant, NSE souffre de limitations d’échantillons dans les études des membranes biologiques. L’un des principaux facteurs limitants de telles expériences est la quantité standard d’échantillon requise pour les mesures NSE (2-4 mL) et les concentrations élevées de l’échantillon s’élevant à 100-200 mg de matériau membranaire (lipides et protéines) pour obtenir des données de haute qualité. Dans de nombreux cas, la production de telles quantités de matériel biologique n’est pas réalisable ou est prohibitive. Dans de tels scénarios, il est possible de réduire la concentration à 20-25 mg/mL, mais cela nécessiterait au moins 4 fois plus de temps d’acquisition afin d’obtenir des statistiques comparables à celles d’échantillons avec des concentrations de 50 mg/mL. Ces exigences strictes en matière de volume et de concentration des échantillons pourraient être allégées avec la prochaine génération de spectromètres NSE sur des sources de neutrons à flux plus élevé, telles que la deuxième station cible du laboratoire national d’Oak Ridge et la source européenne de spallation. Une autre limitation critique dans la réalisation d’expériences NSE sur des membranes lipidiques nécessitant des schémas de deutération sélective est le manque de disponibilité commerciale de certaines variantes deutérées de molécules lipidiques ou leurs prix exorbitants, le cas échéant. Dans certains cas, ces limitations peuvent être contournées en demandant la synthèse de lipides deutérés (ou de cholestérol, protéines) par le biais d’installations de deutération des utilisateurs, telles que le laboratoire de biodération du laboratoire national d’Oak Ridge, l’installation nationale de deutération de l’ANSTO ou l’installation de deutération de la source de neutrons et de muons ISIS. L’accès à ces installations et à leurs capacités de synthèse est possible par le biais de propositions d’utilisateurs soumises qui sont évaluées par des pairs en fonction du mérite scientifique de la synthèse de matériaux proposée et de son utilisation prévue dans les études sensibles aux isotopes.
Malgré ces limites, l’application de la spectroscopie NSE dans les études de la mécanique membranaire a conduit à la détermination des modules de rigidité de flexion des membranes à différents degrés de complexité, des membranes lipidiques monocomposantes35,38 aux membranes biomimétiques multicomposantes41,66,91, qui ont toutes fait progresser notre compréhension de la nature dynamique des membranes lipidiques. Par exemple, les mesures de rigidité de flexion NSE des membranes lipidiques avec différentes unités moléculaires, par exemple, les lipides de différentes longueurs de chaîne acyle et la saturation de la chaîne38,72,92, ont fourni des informations essentielles sur le rôle de la chimie moléculaire dans la mécanique membranaire. Lorsqu’elles sont associées à des informations structurelles, telles que l’épaisseur de la membrane ou l’emballage moléculaire93,ces mesures commencent à fournir de nouvelles perspectives sur l’interdépendance entre la structure et la dynamique de la membrane et sur la façon dont elles influencent la fonction de la membrane. Les échelles mésoscopiques de NSE le positionnent de manière unique pour de telles études fondamentales des relations structure-propriété, les plus pertinentes sur l’échelle de longueur des assemblages moléculaires. Ce sujet a récemment été exploré dans deux études NSE sur les membranes lipidiques riches en cholestérol36 et dans les membranes lipidiques binaires avec inadéquation hydrophobe entre les deux composants lipidiques39. Les deux études ont trouvé des preuves solides que la mécanique membranaire s’adapte à l’aire par lipide, corroborant les conclusions d’une récente simulation de MD entièrement atomique par Doktorova et al.82. Ces résultats soulignent la nature auto-assemblée des membranes lipidiques et fournissent une image unificatrice de l’emballage moléculaire en tant que paramètre clé dans la définition des propriétés dynamiques et fonctionnelles de la membrane.
D’autres applications de la NSE impliquent des études de la réponse mécanique des membranes à de petits additifs, y compris des molécules biologiques telles que lecholestérol 36,37, le tréhalose92et la mélittine73,94, ou des additifs inorganiques tels que les nanoparticules pour les applications d’administration de médicaments76. NSE a également été utilisé pour comprendre comment la mécanique membranaire réagit aux changements dans leur environnement, y compris la température92,le pH74et la présence de macromoléculesd’encombrement 96. De telles études contribuent à une meilleure compréhension des facteurs qui influencent le ramollissement ou le raidissement des membranes lipidiques, dans des conditions biologiques liées à la santé et à la maladie, et dans des contextes contrôlés pour des applications thérapeutiques. Notamment, les mesures NSE ont également été utilisées pour sonder l’effet des peptides antimicrobiens sur la dynamique membranaire73,94,95. D’autres exemples d’applications NSE sur les biomembranes comprennent des études de la dynamique des structures membranaires aplaties, appelées thylakoïdes, qui abritent la machinerie photosynthétique dans les cellules cyanobactériennes97,98.
On peut également utiliser la deutération sélective des lipides dans les études NSE pour étudier la dynamique de caractéristiques membranaires spécifiques qui sont pertinentes pour la fonction biologique. Par exemple, Nickels et al. ont utilisé la deutération lipidique sélective dans les membranes lipidiques formant des domaines pour générer un contraste latéral à l’intérieur de la membrane, comme l’ont déjà illustré Heberle et al.28. Ce schéma de deutération a permis des mesures indépendantes de la rigidité de flexion des domaines lipidiques et de la matrice lipidique hôte41 (voir Figure 2B). Les résultats ont confirmé que les deux compartiments membranaires ont des modules de rigidité de flexion distincts, qui pourraient être un mécanisme moteur pour la formation de domaines dans les membranes cellulaires. Dans une étude plus récente, Rickeard et al. ont utilisé l’échange de cyclodextrine entre liposomes protiés et deutérés pour obtenir des liposomes asymétriques avec des folioles isotopiques40 (Figure 2C). Leurs liposomes finaux avaient une foliole protisée et une foliole deutérée qui est un contraste adapté au tampon, ce qui a permis d’études de la dynamique de la foliole individuelle et de fournir un premier compte rendu expérimental direct de l’effet de l’asymétrie et du couplage de la foliole sur les fluctuations de flexion de la membrane.
La deutération sélective des membranes a également été utilisée dans les études NSE des fluctuations d’épaisseur des membranes, un mode dynamique prédit depuis longtemps dans les membranes lipidiques99 qui n’a été observé que récemment avec l’avènement de la spectroscopie NSE35,100. Ces mesures utilisent des membranes deutérées par la queue pour amplifier le signal provenant des régions du groupe de tête de la membrane et résoudre le signal de fluctuation de l’épaisseur. Ce type d’expériences NSE est relativement récent, mais il a été utilisé efficacement pour comprendre l’interdépendance des propriétés élastiques et visqueuses de la membrane38,pour explorer la mise à l’échelle de la rigidité et de la viscosité de flexion avec l’emballage moléculaire dans les membranes lipidiques mixtes39, et pour sonder les effets locaux du cholestérol sur la viscosité de la membrane36. Un autre domaine d’importance biologique dans lequel ce mode dynamique pourrait avoir des implications de grande portée est les interactions membrane-protéine mésoscopiques95. On sait que la fonction des protéines membranaires est étroitement liée à l’appariement hydrophobe entre la protéine et la membrane hôte. Ainsi, les variations de l’épaisseur de la membrane, dues aux fluctuations d’épaisseur, pourraient agir comme un mécanisme de régulation de la fonction des protéines membranaires. NSE est extrêmement bien adapté à de telles études car il peut sonder directement les effets de la liaison et de l’insertion des protéines sur les fluctuations d’épaisseur de la membrane. Des mesures NSE récentes de notre groupe (non publiées) suggèrent que l’insertion de protéines transmembranaires pourrait supprimer de manière significative les fluctuations d’épaisseur de la membrane et pourrait présenter un mécanisme potentiel pour réguler les événements de signalisation. Il s’agit d’un domaine de recherche urgent, mais sous-développé, où les NSE peuvent avoir un impact significatif sur la compréhension des réponses dynamiques des membranes à la liaison et à l’insertion des protéines sur les échelles de longueur et de temps des fonctions biologiques clés transmises par les interactions des protéines avec les membranes cellulaires.
En résumé, NSE a évolué au cours des dernières années en tant qu’outil puissant pour interroger la dynamique membranaire sur les échelles spatiales et temporelles des fonctions biologiques vitales. La technique suscite rapidement un intérêt généralisé et son potentiel pour répondre à des questions clés sur la fonction membranaire est de plus en plus reconnu. Les capacités de variation de contraste au sein de NSE l’ont positionné comme une approche unique pour mesurer les propriétés membranaires mésoscopiques qui seraient autrement difficiles à obtenir. Un autre avantage significatif de la NSE par rapport aux méthodes de spectroscopie traditionnelles dans les études de la dynamique des membranes est son chevauchement avec les échelles de longueur et de temps accessibles avec les simulations MD, ce qui permet aux études expérimentales / computationnelles synergiques d’acquérir une compréhension au niveau moléculaire des différents composants moléculaires composant les membranes. Malgré ses promesses, l’utilisation de la NSE dans les études sur membrane biologique présente encore certaines limites, notamment l’exigence de grands volumes d’échantillons, la difficulté de la deutération sélective dans les systèmes biologiques et le flux de neutrons relativement faible sur les spectromètres NSE, ce qui se traduit par des temps de mesure plus longs et une disponibilité limitée du temps de faisceau. Cependant, ces lacunes pourraient être surmontées dans un proche avenir avec des développements constants dans les sources de neutrons et l’instrumentation ainsi que des progrès dans les installations de deutération.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs ne déclarent aucun conflit d’intérêts et n’ont rien à divulguer.
Acknowledgments
R. Ashkar remercie M. Nagao, L.-R. Stingaciu et P. Zolnierczuk pour de nombreuses discussions utiles et pour leur aide fréquente dans les expériences NSE sur leurs lignes de faisceau respectives. Les auteurs reconnaissent l’utilisation de spectromètres d’écho de spin neutronique au NIST et à l’ORNL. Le spectromètre NSE du NIST est soutenu par le Center for High Resolution Neutron Scattering, un partenariat entre le National Institute of Standards and Technology et la National Science Foundation en vertu de l’accord no. DMR-1508249. Le spectromètre NSE de la source de neutrons de spallation de l’ORNL est soutenu par la Division des installations pour les utilisateurs scientifiques, Office of Basic Energy Sciences, Département de l’énergie des États-Unis. Oak Ridge National Laboratory est géré par UT-Battelle, LLC en vertu du contrat US DOE No. DE-AC05-00OR22725.
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform (biotech grade) | Sigma Aldrich | 496189 | Biotech. grade, ≥99.8%, contains 0.5-1.0% ethanol as stabilizer |
| Circulating water bath | Julabo | SE-12 | Heating Circulator with smart pump, programmable temperature settings, and external sensor connection for measurement and control |
| Deuterium Oxide | Cambridge Isotopes Laboratories | DLM-4 | Deuterated water; Heavy water (D2O) (D, 99.9%) |
| Digital Semi-Microbalance | Mettler Toledo | MS105 | Semi-micro balance with 120 g capacity, 0.01 mg readability, high resolution weighing cell, ergonomic doors, and pipette-check application |
| Ethanol (molecular biology grade) | Sigma Aldrich | E7023 | 200 proof ethanol for molecular biology applications |
| Glass Pipets | VWR | 36360-536 | Disposable Soda Lime glass Pasteur pipets |
| Glass Vials | Thermo Scientific | B7990-1 | Borosilicate glass vials with PTFE/Silione septum caps |
| Lab grade freezer | Fisher Scientific | IU2886D | Ultra-low temprature freezer (-86 to -50 C) for long-term storage of lipids and proteins |
| Lipids (protaited or perdeuterated) | Avanti Polar Lipids | varies by lipid | Lipids can be purchased from Avanti in powder form or in a chloroform solution with the required amounts and deuteration schemes. |
| Millipore water purifier | Millipore Sigma | ZRQSVP3US | Direct-Q® 3 UV Water Purification System which deliver both pure and ultrapure water with a built-in UV lamp to reduce the levels of organics for biological applications |
| Mini Extruder Set | Avanti Polar Lipids | 610020 | Mini-extruder set includes mini-extruder, heating block, 2 GasTight Syringes, and 2 O-rings, Polycarbonate Membranes, and Filter Supports |
| Quick Connect Fittings | Grainger | 2YDA1 and 2YDA7 | Push-button tube fittings for QuickConnect water circulation applications, e.g. high temperature vesicle extrusion |
| Syringe Pump | SyringePump.com | New Era-1000 | Fully programmable syringe pump for infusion and withdrawal; programs up to 41 pumping phases with adjustable pumping rates, dispensed volumes, and extrusion cycles |
| Ultrasonic bath | Fisher Scientific | CPX2800 | Temperature controlled ultra sonic bath with programmable functionality for degassing and ultrasonic applications |
| Vacuum Oven | Thermo Scientific | 3608 | 0.7 cu ft vaccum oven with built-in-high-limit thermostat guards against overheating |
| Vortex Mixer | Fisher Scientific | 02-215-414 | Variable speed, analog control that allows low rpm start-up for gentle shaking or high-speed mixing for vigorous vortexing of samples |
References
- Singer, S. J., Nicolson, G. L. The fluid mosaic model of the structure of cell membranes. Science. 175 (4023), 720-731 (1972).
- Andersen, O. S., Koeppe, R. E. Bilayer thickness and membrane protein function: an energetic perspective. Annual Review of Biophysics and Biomolecular Structure. 36, 107-130 (2007).
- Lundbæk, J. A., Collingwood, S. A., Ingólfsson, H. I., Kapoor, R., Andersen, O. S. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. Journal of The Royal Society Interface. 7 (44), 373-395 (2010).
- Bradley, R. P., Radhakrishnan, R. Curvature-undulation coupling as a basis for curvature sensing and generation in bilayer membranes. Proceedings of the National Academy of Sciences of the United States of America. 113 (35), 117-124 (2016).
- Perozo, E., Cortes, D. M., Sompornpisut, P., Kloda, A., Martinac, B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418 (6901), 942-948 (2002).
- Jensen, M. Ø, Mouritsen, O. G. Lipids do influence protein function-the hydrophobic matching hypothesis revisited. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1666 (1-2), 205-226 (2004).
- Rajendran, L., Simons, K. Lipid rafts and membrane dynamics. Journal of Cell Science. 118 (6), 1099-1102 (2005).
- Katchalsky, A., Spangler, R. Dynamics of membrane processes. Quarterly Reviews of Biophysics. 1 (2), 127-175 (1968).
- Rheinstädter, M. C. Collective molecular dynamics in proteins and membranes (Review). Biointerphases. 3 (2), 83-90 (2008).
- Fujiwara, T., Ritchie, K., Murakoshi, H., Jacobson, K., Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. The Journal of Cell Biology. 157 (6), 1071-1082 (2002).
- Hac, A. E., Seeger, H. M., Fidorra, M., Heimburg, T. Diffusion in two-component lipid membranes--a fluorescence correlation spectroscopy and monte carlo simulation study. Biophysical Journal. 88 (1), 317-333 (2005).
- Heinrich, M., Tian, A., Esposito, C., Baumgart, T. Dynamic sorting of lipids and proteins in membrane tubes with a moving phase boundary. Proceedings of the National Academy of Sciences of the United States of America. 107 (16), 7208-7213 (2010).
- Hormel, T. T., Kurihara, S. Q., Brennan, M. K., Wozniak, M. C., Parthasarathy, R. Measuring lipid membrane viscosity using rotational and translational probe diffusion. Physical Review Letters. 112 (18), 188101 (2014).
- Dimova, R. Recent developments in the field of bending rigidity measurements on membranes. Advances in Colloid and Interface Science. 208, 225-234 (2014).
- Bassereau, P., Sorre, B., Lévy, A. Bending lipid membranes: Experiments after W. Helfrich's model. Advances in Colloid and Interface Science. 208, 47-57 (2014).
- Monzel, C., Sengupta, K. Measuring shape fluctuations in biological membranes. Journal of Physics D: Applied Physics. 49 (24), 243002 (2016).
- Deserno, M. Mesoscopic membrane physics: concepts, simulations, and selected applications. Macromolecular Rapid Communications. 30 (9-10), 752-771 (2009).
- Reynwar, B. J., et al. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 447 (7143), 461-464 (2007).
- Haswell, E. S., Phillips, R., Rees, D. C. Mechanosensitive channels: what can they do and how do they do it. Structure. 19 (10), 1356-1369 (2011).
- Phillips, R., Ursell, T., Wiggins, P., Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature. 459 (7245), 379-385 (2009).
- Dill, K. A., Chan, H. S. From Levinthal to pathways to funnels. Nature Structural Biology. 4 (1), 10-19 (1997).
- Henzler-Wildman, K., Kern, D. Dynamic personalities of proteins. Nature. 450 (7172), 964-972 (2007).
- Grimaldo, M., Roosen-Runge, F., Zhang, F., Schreiber, F., Seydel, T. Dynamics of proteins in solution. Quarterly Reviews of Biophysics. 52, 7 (2019).
- Lyman, E., Hsieh, C. -L., Eggeling, C. From dynamics to membrane organization: experimental breakthroughs occasion a "modeling manifesto". Biophysical Journal. 115 (4), 595-604 (2018).
- Arriaga, L. R., et al. Dissipative curvature fluctuations in bilayer vesicles: Coexistence of pure-bending and hybrid curvature-compression modes. The European Physical Journal. E, Soft Matter. 31 (1), 105-113 (2010).
- Honerkamp-Smith, A. R., Veatch, S. L., Keller, S. L. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1788 (1), 53-63 (2009).
- Veatch, S. L., Keller, S. L. Organization in lipid membranes containing cholesterol. Physical Review Letters. 89 (26), 268101 (2002).
- Heberle, F. A., et al. Bilayer thickness mismatch controls domain size in model membranes. Journal of the American Chemical Society. 135 (18), 6853-6859 (2013).
- Nickels, J. D., et al. The in vivo structure of biological membranes and evidence for lipid domains. PLOS Biology. 15 (5), 2002214 (2017).
- Simons, K., Ikonen, E. Functional rafts in cell membranes. Nature. 387 (6633), 569-572 (1997).
- van Meer, G., Voelker, D. R., Feigenson, G. W. Membrane lipids: where they are and how they behave. Nature Reviews. Molecular Cell Biology. 9 (2), 112-124 (2008).
- Liu, S. -L., et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nature Chemical Biology. 13, 268 (2016).
- Rothman, J., Lenard, J. Membrane asymmetry. Science. 195 (4280), 743-753 (1977).
- Ashkar, R., et al. Neutron scattering in the biological sciences: progress and prospects. Acta Crystallographica Section D. 74 (12), 1129-1168 (2018).
- Woodka, A. C., Butler, P. D., Porcar, L., Farago, B., Nagao, M. Lipid bilayers and membrane dynamics: insight into thickness fluctuations. Physical Review Letters. 109 (5), 058102 (2012).
- Chakraborty, S., et al. How cholesterol stiffens unsaturated lipid membranes. Proceedings of the National Academy of Sciences of the United States of America. 117 (36), 21896-21905 (2020).
- Arriaga, L. R., et al. Stiffening effect of cholesterol on disordered lipid phases: a combined neutron spin echo + dynamic light scattering analysis of the bending elasticity of large unilamellar vesicles. Biophysical Journal. 96 (9), 3629-3637 (2009).
- Nagao, M., Kelley, E. G., Ashkar, R., Bradbury, R., Butler, P. D. Probing elastic and viscous properties of phospholipid bilayers using neutron spin echo spectroscopy. The Journal of Physical Chemistry Letters. 8 (19), 4679-4684 (2017).
- Kelley, E. G., Butler, P. D., Ashkar, R., Bradbury, R., Nagao, M. Scaling relationships for the elastic moduli and viscosity of mixed lipid membranes. Proceedings of the National Academy of Sciences of the United States of America. 117 (38), 23365-23373 (2020).
- Rickeard, B. W., et al. Transverse lipid organization dictates bending fluctuations in model plasma membranes. Nanoscale. 12 (3), 1438-1447 (2020).
- Nickels, J. D., et al. Mechanical properties of nanoscopic lipid domains. Journal of the American Chemical Society. 137 (50), 15772-15780 (2015).
- Mezei, F. Neutron spin echo: A new concept in polarized thermal neutron techniques. Zeitschrift für Physik A Hadrons and Nuclei. 255 (2), 146-160 (1972).
- Hayter, J. B., Penfold, J. Neutron spin-echo integral transform spectroscopy. Zeitschrift für Physik B Condensed Matter. 35 (2), 199-205 (1979).
- Monkenbusch, M., Richter, D. Neutrons in Soft Matter. Imae, T., Kanaya, T., Furusaka, M., Torikai, N. , Wiley. ch6 147-182 (2011).
- Pynn, R. Neutron Spin Echo. Mezei, F., Pappas, C., Gutberlet, T. , Springer. Berlin Heidelberg. 159-177 (2003).
- Holderer, O., et al. The JCNS neutron spin-echo spectrometer J-NSE at the FRM II. Measurement Science and Technology. 19 (3), 034022 (2008).
- Schleger, P., et al. The long-wavelength neutron spin-echo spectrometer IN15 at the Institut Laue-Langevin. Physica B: Condensed Matter. 241-243, 164-165 (1997).
- Holderer, O., Zolnierczuk, P., Pasini, S., Stingaciu, L., Monkenbusch, M. A better view through new glasses: Developments at the Jülich neutron spin echo spectrometers. Physica B: Condensed Matter. 562, 9-12 (2019).
- Farago, B., et al. The IN15 upgrade. Neutron News. 26 (3), 15-17 (2015).
- Ashkar, R. Selective dynamics in polymeric materials: Insights from quasi-elastic neutron scattering spectroscopy. Journal of Applied Physics. 127 (15), 151101 (2020).
- Pasini, S., Holderer, O., Kozielewski, T., Richter, D., Phoenix Monkenbusch, M. J-NSE- Phoenix, a neutron spin-echo spectrometer with optimized superconducting precession coils at the MLZ in Garching. Review of Scientific Instruments. 90 (4), 043107 (2019).
- Svergun, D. I., Koch, M. H. J., Timmins, P. A., May, R. P. Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules. , Oxford University Press. (2013).
- Eicher, B., et al. Joint small-angle X-ray and neutron scattering data analysis of asymmetric lipid vesicles. Journal of Applied Crystallography. 50 (2), 419-429 (2017).
- Heberle, F. A., et al. Model-based approaches for the determination of lipid bilayer structure from small-angle neutron and X-ray scattering data. European Biophysics Journal. 41 (10), 875-890 (2012).
- Jaksch, S., Koutsioubas, A., Mattauch, S., Holderer, O., Frielinghaus, H. Long-range excitations in phospholipid membranes. Chemistry and Physics of Lipids. 225, 104788 (2019).
- Jaksch, S., et al. Influence of ibuprofen on phospholipid membranes. Physical Review E. 91 (2), 022716 (2015).
- Armstrong, C. L., et al. Effect of cholesterol on the lateral nanoscale dynamics of fluid membranes. European Biophysics Journal. 41 (10), 901-913 (2012).
- Rheinstädter, M. C., Häußler, W., Salditt, T. Dispersion relation of lipid membrane shape fluctuations by neutron spin-echo spectrometry. Physical Review Letters. 97 (4), 048103 (2006).
- Armstrong, C. L., Häußler, W., Seydel, T., Katsaras, J., Rheinstädter, M. C. Nanosecond lipid dynamics in membranes containing cholesterol. Soft Matter. 10 (15), 2600-2611 (2014).
- Nickels, J. D., et al. Lipid rafts: buffers of cell membrane physical properties. The Journal of Physical Chemistry B. 123 (9), 2050-2056 (2019).
- Michonova-Alexova, E. I., Sugár, I. P. Component and state separation in DMPC/DSPC lipid bilayers: a Monte Carlo simulation study. Biophysical Journal. 83 (4), 1820-1833 (2002).
- Sugár, I. P., Thompson, T. E., Biltonen, R. L. Monte Carlo simulation of two-component bilayers: DMPC/DSPC mixtures. Biophysical Journal. 76 (4), 2099-2110 (1999).
- Mabrey, S., Sturtevant, J. M. Investigation of phase transitions of lipids and lipid mixtures by sensitivity differential scanning calorimetry. Proceedings of the National Academy of Sciences. 73 (11), 3862-3866 (1976).
- Neutron activation and scattering calculator. , Available from: https://www.ncnr.nist.gov/resources/activation/ (2021).
- Scott, H. L., et al. On the mechanism of bilayer separation by extrusion, or why your LUVs are not really unilamellar. Biophysical Journal. 117 (8), 1381-1386 (2019).
- Ashkar, R., et al. Tuning membrane thickness fluctuations in model lipid bilayers. Biophysical Journal. 109 (1), 106-112 (2015).
- Carrillo, J. -M. Y., Katsaras, J., Sumpter, B. G., Ashkar, R. A computational approach for modeling neutron scattering data from lipid bilayers. Journal of Chemical Theory and Computation. 13 (2), 916-925 (2017).
- Azuah, R. T. DAVE: a comprehensive software suite for the reduction, visualization, and analysis of low energy neutron spectroscopic data. Journal of Research of the National Institute of Standards and Technology. 114 (6), 341-358 (2009).
- Van Hove, L. Correlations in space and time and born approximation scattering in systems of interacting particles. Physical Review. 95 (1), 249-262 (1954).
- Zilman, A. G., Granek, R. Undulations and dynamic structure factor of membranes. Physical Review Letters. 77 (23), 4788-4791 (1996).
- Kelley, E. G., Butler, P. D., Nagao, M. Collective dynamics in model biological membranes measured by neutron spin echo spectroscopy. , Walter de Gruyter, Inc. 131-176 (2019).
- Zheng, Y., Michihiro, N., Dobrin, P. B. Bending elasticity of saturated and monounsaturated phospholipid membranes studied by the neutron spin echo technique. Journal of Physics: Condensed Matter. 21 (15), 155104 (2009).
- Sharma, V. K., Qian, S. Effect of an antimicrobial peptide on lateral segregation of lipids: a structure and dynamics study by neutron scattering. Langmuir. 35 (11), 4152-4160 (2019).
- Boggara, M. B., Faraone, A., Krishnamoorti, R. Effect of pH and Ibuprofen on the Phospholipid Bilayer Bending Modulus. The Journal of Physical Chemistry B. 114 (24), 8061-8066 (2010).
- Lee, J. -H., et al. Thermal fluctuation and elasticity of lipid vesicles interacting with pore-forming peptides. Physical Review Letters. 105 (3), 038101 (2010).
- Chakraborty, S., Abbasi, A., Bothun, G. D., Nagao, M., Kitchens, C. L. Phospholipid bilayer softening due to hydrophobic gold nanoparticle inclusions. Langmuir. 34 (44), 13416-13425 (2018).
- Hoffmann, I., et al. Softening of phospholipid membranes by the adhesion of silica nanoparticles - as seen by neutron spin-echo (NSE). Nanoscale. 6 (12), 6945-6952 (2014).
- Watson, M. C., Brown, F. L. H. Interpreting membrane scattering experiments at the mesoscale: the contribution of dissipation within the bilayer. Biophysical Journal. 98 (6), 9-11 (2010).
- Seifert, U., Langer, S. A. Viscous modes of fluid bilayer membranes. Europhysics Letters (EPL). 23 (1), 71-76 (1993).
- Bingham, R. J., Smye, S. W., Olmsted, P. D. Dynamics of an asymmetric bilayer lipid membrane in a viscous solvent. EPL (Europhysics Letters). 111 (1), 18004 (2015).
- Rawicz, W., Olbrich, K. C., McIntosh, T., Needham, D., Evans, E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophysical Journal. 79 (1), 328-339 (2000).
- Doktorova, M., LeVine, M. V., Khelashvili, G., Weinstein, H. A new computational method for membrane compressibility: bilayer mechanical thickness revisited. Biophysical Journal. 116 (3), 487-502 (2019).
- Evans, E., Needham, D. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion and colloidal interactions. The Journal of Physical Chemistry. 91 (16), 4219-4228 (1987).
- Lesieur, S., Grabielle-Madelmont, C., Paternostre, M. T., Ollivon, M. Size analysis and stability study of lipid vesicles by high-performance gel exclusion chromatography, turbidity, and dynamic light scattering. Analytical Biochemistry. 192 (2), 334-343 (1991).
- Heberle, F. A., et al. Direct label-free imaging of nanodomains in biomimetic and biological membranes by cryogenic electron microscopy. Proceedings of the National Academy of Sciences of the United States of America. 117 (33), 19943-19952 (2020).
- Cornell, C. E., Mileant, A., Thakkar, N., Lee, K. K., Keller, S. L. Direct imaging of liquid domains in membranes by cryo-electron tomography. Proceedings of the National Academy of Sciences of the United States of America. 117 (33), 19713-19719 (2020).
- Yao, X., Fan, X., Yan, N. Cryo-EM analysis of a membrane protein embedded in the liposome. Proceedings of the National Academy of Sciences of the United States of America. 117 (31), 18497-18503 (2020).
- Kučerka, N., Nieh, M. -P., Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1808 (11), 2761-2771 (2011).
- Nielsen, J. E., Bjørnestad, V. A., Lund, R. Resolving the structural interactions between antimicrobial peptides and lipid membranes using small-angle scattering methods: the case of indolicidin. Soft Matter. 14 (43), 8750-8763 (2018).
- Kučerka, N., et al. Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophysical Journal. 95 (5), 2356-2367 (2008).
- Kelley, E. G., Butler, P. D., Nagao, M. Scaling of lipid membrane rigidity with domain area fraction. Soft Matter. 15 (13), 2762-2767 (2019).
- Brüning, B. -A., et al. Bilayer undulation dynamics in unilamellar phospholipid vesicles: Effect of temperature, cholesterol and trehalose. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1838 (10), 2412-2419 (2014).
- Kučerka, N., et al. Areas of monounsaturated diacylphosphatidylcholines. Biophysical Journal. 97 (7), 1926-1932 (2009).
- Sharma, V. K., Mamontov, E., Anunciado, D. B., O'Neill, H., Urban, V. S. Effect of antimicrobial peptide on the dynamics of phosphocholine membrane: role of cholesterol and physical state of bilayer. Soft Matter. 11 (34), 6755-6767 (2015).
- Kelley, E. G., Butler, P. D., Nagao, M. Collective dynamics in lipid membranes containing transmembrane peptides. Soft Matter. , (2021).
- Yu, J., et al. Structure and dynamics of lipid membranes interacting with antivirulence end-phosphorylated polyethylene glycol block copolymers. Soft Matter. 16 (4), 983-989 (2020).
- Stingaciu, L. -R., et al. Revealing the dynamics of thylakoid membranes in living cyanobacterial cells. Scientific Reports. 6 (1), 19627 (2016).
- Stingaciu, L. -R., O'Neill, H. M., Liberton, M., Pakrasi, H. B., Urban, V. S. Influence of chemically disrupted photosynthesis on cyanobacterial thylakoid dynamics in synechocystis sp. PCC 6803. Scientific Reports. 9 (1), 5711 (2019).
- Miller, I. R. Energetics of fluctuation in lipid bilayer thickness. Biophysical Journal. 45 (3), 643-644 (1984).
- Nagao, M. Observation of local thickness fluctuations in surfactant membranes using neutron spin echo. Physical Review E. 80 (3), 031606 (2009).
Tags
Biologie numéro 171 dynamique des lipides collectifs module de flexion compressibilité de surface fluctuations d’épaisseur viscosité membranaire deutération sélective préparation de liposomesErratum
Formal Correction: Erratum: Neutron Spin Echo Spectroscopy as a Unique Probe for Lipid Membrane Dynamics and Membrane-Protein Interactions
Posted by JoVE Editors on 08/06/2021.
Citeable Link.
An erratum was issued for: Neutron Spin Echo Spectroscopy as a Unique Probe for Lipid Membrane Dynamics and Membrane-Protein Interactions. The Introduction, Protocol, and Representative Results sections have been updated.
In the Introduction, the fith pargraph was updated from:
Besides direct access to the length and time scale of membrane dynamics, NSE has the inherent capabilities of neutron isotope sensitivity52. Specifically, the ability of neutrons to interact differently with the isotopes of hydrogen, the most abundant element in biological systems, results in a different neutron scattering length density,34 or NSLD (the equivalent of the optical index of refraction50), when protium is substituted by deuterium. This enables an approach known as contrast variation, which is commonly used to highlight specific membrane features or conceal others — the latter scenario is referred to as contrast matching. A frequent application of contrast variation/matching is the substitution of water (NSLD = -0.56 × 10-6 Å-2) by heavy water or D2O (NSLD = 6.4 × 10-6 Å-2) to amplify the neutron signal from protiated lipid membranes (NSLD ~ 2 × 10-6 Å-2). This approach is highly effective in studies of membrane structure because the penetration of D2O into the headgroup region of the membrane allows accurate determination of the membrane thicknesses (see Figure 2A, left panel) and of the location of different lipid subgroups when more sophisticated models are applied53,54. This paper highlights some examples on the use of contrast variation for studies of collective dynamics in biomimetic membranes and select membrane features.
to:
Besides direct access to the length and time scale of membrane dynamics, NSE has the inherent capabilities of neutron isotope sensitivity52. Specifically, the ability of neutrons to interact differently with the isotopes of hydrogen, the most abundant element in biological systems, results in a different neutron scattering length density,34 or NSLD (the equivalent of the optical index of refraction50), when protium is substituted by deuterium. This enables an approach known as contrast variation, which is commonly used to highlight specific membrane features or conceal others — the latter scenario is referred to as contrast matching. A frequent application of contrast variation/matching is the substitution of water (NSLD = -0.56 × 10-6 Å-2) by heavy water or D2O (NSLD = 6.4 × 10-6 Å-2) to amplify the neutron signal from protiated lipid membranes (NSLD ~ 0 × 10-6 Å-2). This approach is highly effective in studies of membrane structure because the penetration of D2O into the headgroup region of the membrane allows accurate determination of the membrane thicknesses (see Figure 2A, left panel) and of the location of different lipid subgroups when more sophisticated models are applied53,54. This paper highlights some examples on the use of contrast variation for studies of collective dynamics in biomimetic membranes and select membrane features.
In the Protocol, step 1.1 was updated from:
For bending fluctuation measurements, make fully protiated liposomes in D2O (D 99.9%) or D2O-buffer (e.g., phosphate buffer prepared with D2O instead of H2O). Use fully protiated DMPC (C36H72NO8P) and DSPC (C44H88NO8P) with 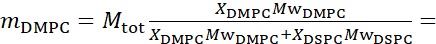 133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 2 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 2 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
to:
For bending fluctuation measurements, make fully protiated liposomes in D2O (D 99.9%) or D2O-buffer (e.g., phosphate buffer prepared with D2O instead of H2O). Use fully protiated DMPC (C36H72NO8P) and DSPC (C44H88NO8P) with 133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 0 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
In the Representative Results, the fist pagargaph was updted from:
NSE studies accessing bending fluctuations are typically performed over a Q-range of ~ (0.04 - 0.2) Å-1. This Q-range corresponds to intermediate length scales between the membrane thickness and the liposomal radius, where bending dynamics dominate. Measurement over an extended Q-range can give access to additional dynamic modes, including liposomal diffusion and intramembrane dynamics. For more details on the cross-over in membrane dynamics accessed by NSE, check these relevant publications25,71. It is important to emphasize that NSE signals are proportional to: 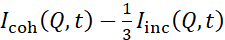 , where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~2 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
, where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~2 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
to:
NSE studies accessing bending fluctuations are typically performed over a Q-range of ~ (0.04 - 0.2) Å-1. This Q-range corresponds to intermediate length scales between the membrane thickness and the liposomal radius, where bending dynamics dominate. Measurement over an extended Q-range can give access to additional dynamic modes, including liposomal diffusion and intramembrane dynamics. For more details on the cross-over in membrane dynamics accessed by NSE, check these relevant publications25,71. It is important to emphasize that NSE signals are proportional to: , where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~0 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
In the Representative Reults, Figure 2 was updated from:
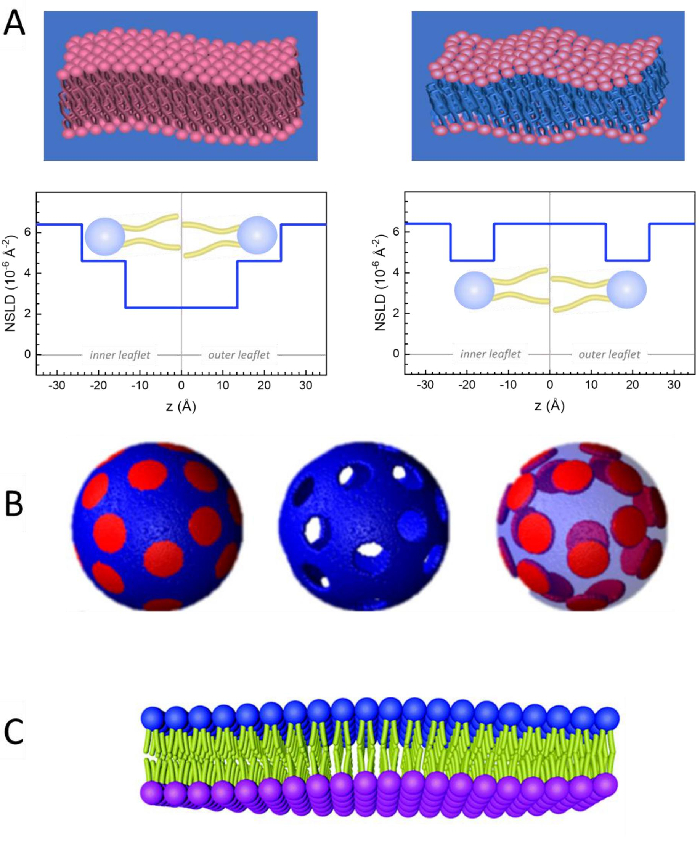
Figure 2: Examples of possible deuteration schemes in NSE experiments on lipid membranes. (A) Left: Fully contrasted membranes, e.g., protiated membranes in deuterated buffer, showing the NSLD profile along the normal to the membrane surface. The difference in the NSLD between the headgroup (~2 × 10-2 Å-2) and tail region (~4.5 × 10-6 Å-2) of the membrane is due to the headgroup hydration with deuterated buffer. Right: Tail-contrast matched membranes such that the hydrocarbon tail region of the membrane has the same NSLD as the buffer, as shown in the corresponding NSLD profile along the membrane normal. (B) Domain-forming membranes with two neutron contrast schemes where the domains (center) or the matrix (left) are contrast-matched to the buffer, enabling selective studies of matrix or domain dynamics, respectively. This figure has been modified from Nickels et al., JACS 201541. (C) Asymmetric membranes prepared by cyclodextrin exchange between protiated and deuterated lipid vesicles, resulting in the deuteration of one membrane leaflet while keeping the other leaflet protiated. This allows studies of the bending dynamics of the protiated leaflet and provides insights into the mechanical coupling between opposing leaflets in asymmetric membranes. This figure has been modified from Rickeard et al., Nanoscale 202040. Please click here to view a larger version of this figure.
to:
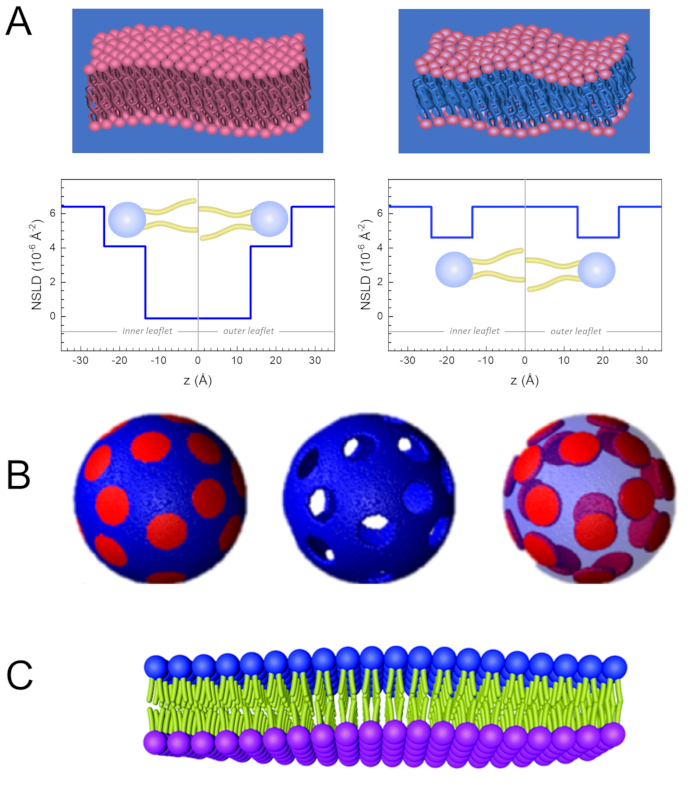
Figure 2: Examples of possible deuteration schemes in NSE experiments on lipid membranes. (A) Left: Fully contrasted membranes, e.g., protiated membranes in deuterated buffer, showing the NSLD profile along the normal to the membrane surface. The difference in the NSLD between the tail region (~0 × 10-2 Å-2) and headgroup region (~4.5 × 10-6 Å-2) of the membrane is due to the headgroup hydration with deuterated buffer. Right: Tail-contrast matched membranes such that the hydrocarbon tail region of the membrane has the same NSLD as the buffer, as shown in the corresponding NSLD profile along the membrane normal. (B) Domain-forming membranes with two neutron contrast schemes where the domains (center) or the matrix (left) are contrast-matched to the buffer, enabling selective studies of matrix or domain dynamics, respectively. This figure has been modified from Nickels et al., JACS 201541. (C) Asymmetric membranes prepared by cyclodextrin exchange between protiated and deuterated lipid vesicles, resulting in the deuteration of one membrane leaflet while keeping the other leaflet protiated. This allows studies of the bending dynamics of the protiated leaflet and provides insights into the mechanical coupling between opposing leaflets in asymmetric membranes. This figure has been modified from Rickeard et al., Nanoscale 202040. Please click here to view a larger version of this figure.
