

Abstract
Cancer is currently the second most common cause of death worldwide. The hallmark of cancer cells is the presence of specific marker proteins such as growth factor receptors on their surface. This feature enables development of highly selective therapeutics, the protein bioconjugates, composed of targeting proteins (antibodies or receptor ligands) connected to highly cytotoxic drugs by a specific linker. Due to very high affinity and selectivity of targeting proteins the bioconjugates recognize marker proteins on the cancer cells surface and utilize receptor-mediated endocytosis to reach the cell interior. Intracellular vesicular transport system ultimately delivers the bioconjugates to the lysosomes, where proteolysis separates free cytotoxic drugs from the proteinaceous core of the bioconjugates, triggering drug-dependent cancer cell death. Currently, there are several protein bioconjugates approved for cancer treatment and large number is under development or clinical trials.
One of the main challenges in the generation of the bioconjugates is a site-specific attachment of the cytotoxic drug to the targeting protein. Recent years have brought a tremendous progress in the development of chemical and enzymatic strategies for protein modification with cytotoxic drugs. Here we present the detailed protocols for the site-specific incorporation of cytotoxic warheads into targeting proteins using a chemical method employing maleimide-thiol chemistry and an enzymatic approach that relies on sortase A-mediated ligation. We use engineered variant of fibroblast growth factor 2 and fragment crystallizable region of human immunoglobulin G as an exemplary targeting proteins and monomethyl auristatin E and methotrexate as model cytotoxic drugs. All the described strategies allow for highly efficient generation of biologically active cytotoxic conjugates of defined molecular architecture with potential for selective treatment of diverse cancers.
Introduction
Decades of scientific efforts have led to an enormous advancement in our knowledge about the molecular mechanisms governing cancer development and progression. At the same time, the therapeutic possibilities are still largely limited due to the adverse effects of drugs caused by their lack of selectivity, the great variability of tumors and drug-resistance developed after prolonged treatment. Targeted anti-cancer therapies have been gaining attention in recent years as novel and highly promising approaches for treatment of diverse tumors. Targeted therapies rely on sophisticated drug delivery systems that precisely deliver the cytotoxic payload to the cancer cells and spare the healthy ones. These include mainly diverse nanoparticles, liposomes, and protein-based drug carriers.
Cancer cells often expose elevated levels of specific marker proteins on their surface. Antibody drug conjugates (ADCs) are novel protein-based anti-cancer therapeutics, which combine in one molecule extreme specificity of monoclonal antibodies and high cytotoxic potency of drugs. Once bound to the cancer cell surface, ADC utilize receptor-mediated endocytosis to enter the cell. Subsequently, ADCs are transported via endosomal compartments to the lysosomes, where proteases degrade ADCs and release active cytotoxic drugs. Currently, there are eight ADCs approved in the US for the treatment of diverse tumors, including triple negative breast cancer, HER2 positive breast cancer, urothelial cancer, diffuse large B-cell lymphoma, hematological malignancy, Hodgkin lymphoma and acute myeloid leukemia. Large number of ADCs are also either under development or await approval1. Noteworthy, protein engineering approaches have led to the development of diverse alternative to monoclonal antibodies protein scaffolds and their cytotoxic conjugates. These include different antibody fragments2,3, DARPins4,5, knottins6,7, centyrins8, affibodies9,10, or engineered receptor ligands11,12.
There are several critical requirements that have to be met by a successful protein-based cytotoxic conjugate, namely the conjugate stability, extraordinary specificity, high affinity of the conjugate towards cancer-specific marker, rapid internalization of the conjugate into the cancer cell interior, its efficient transport to the lysosomes and effective intracellular release of the active payload. Another important feature is conjugates homogeneity, which largely depends on the applied strategy for the attachment of the payload to the targeting proteins. There are several methods available for site-specific conjugation of proteins with cytotoxic drugs, like modification of protein side-chain cysteine or lysine residues, attachment of the drug to unnatural amino acids incorporated into the targeting proteins, or enzymatic modifications of the targeting proteins (e.g., with transglutaminase, glycosyltransferase, formylglycine-generating enzyme, sortase A). In most cases site-specific conjugation methods require modifications of the targeting molecules (e.g., via cysteine engineering or introduction of short peptide tags), but in turn result in an efficient production of homogenous conjugate of interest.
Here we provide protocols for highly efficient site-specific conjugation of targeting proteins with cytotoxic drugs. As exemplary proteins we used two different molecules: the fragment crystallizable (Fc) of human IgG and an engineered variant of human fibroblast growth factor 2 (FGF2). The Fc fragment constitutes integral part of typical ADCs, but it is also present in other types of conjugates like cytotoxic peptibodies or conjugates of antibody fragments. FGF2 is a natural fibroblast growth factor receptor (FGFR) ligand that was successfully engineered to yield a selective cytotoxic conjugate targeting FGFR-overproducing cancer cells.
We present two distinct conjugation strategies allowing for the site-specific incorporation of cytotoxic drugs. First, the protocol for conjugation to the cysteine side chains of the Fc fragment via maleimide-thiol chemistry based on Hermanson’s protocol13 is provided (Figure 1A,B). In this protocol two disulfide linkages are initially reduced with tris(2-carboxyethyl)phosphine (TCEP) and resulting free thiol groups are subjected to conjugation with monomethyl auristatin E (MMAE) via maleimide-thiol chemistry (Figure 1B). Due to the interaction between constant heavy chain domains 2 and 3 (CH2 and CH3) the dimeric structure of the drug-linked Fc is preserved. Secondly, the strategy for the generation of double warhead FGF2 conjugate is presented that combines cysteine engineering and sortase A-mediated ligation for incorporation of two distinct drugs into FGF2 in a site-specific manner (Figure 1A,C). The cysteine-free variant of FGF2 bearing additional N-terminal KCKSGG with single exposed cysteine residue and C-terminal LPETGG short peptide tag is used12. The maleimide-thiol reaction allows for the conjugation of MMAE to cysteine within KCKSSG linker designed by our group14,15. Sortase A-dependent step (based on Chen et al.)16 mediates the ligation of methotrexate (MTX)-linked tetraglycine peptide GGGG-MTX to the C-terminal LPETGG sequence, yielding two types of single warhead conjugates (Figure 1C). Sortase A is a cysteine protease that catalyzes the transpeptidation reaction between LPETGG and GGGG motifs. The enzyme binds to the LPETGG motif at the C-terminus of the protein, then the amide bond between the threonine and the glycine is hydrolyzed to form an enzyme-substrate complex. The next step is the aminolysis of the thioester enzyme-substrate bond, where the donor of a primary amino group is the glycine residue of the tetraglycine motif17. Combination of these two approaches generates site-specific FGF2 double warhead conjugates (Figure 1C). In principle, provided conjugation protocols can be successfully applied to any engineered targeting protein of interest to generate selective cytotoxic conjugates. Moreover, versatility of this approach makes it suitable for many other protein-protein and protein-peptide ligation purposes, as well as for the attachment of lipids, polymers, nucleic acids and fluorophores to proteins with available sulfhydryl group (or generated by reduction of native disulfide bonds) and/or with introduced small peptide tag.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Conjugation of the Fc-domain with MMAE
NOTE: Prior to site-specific conjugations prepare key reagents: highly pure protein of interest (in this case the Fc fragment and engineered FGF2 variant, as a starting point 1-5 mg of recombinant protein, prepared according to Sokolowska-Wedzina18), maleimidocaproyl-Val-Cit-p-aminobenzyl alcohol (PABC)-monomethyl auristatin E (MMAE) (CAUTION, highly cytotoxic agent, handle with care), Tris(2-carboxyethyl)phosphine (TCEP) and sortase A16. Methotrexate linked to tetraglycine peptide (GGGG-MTX) (CAUTION, MTX is a highly cytotoxic agent, handle with care) can be either synthesized on the solid support phase according to the solid phase peptide synthesis (SPPS) method in the Fmoc strategy19 or obtained from commercial sources.
- To reduce disulfide bonds within the Fc fragment (Figure 1), add tenfold molar excess of TCEP over the Fc protein (39 µM) in PBS (100 mM NaCl, 18 mM NaH2PO4, 33 mM Na2HPO4 pH 7.4) and incubate at 37 ˚C for 1 h. TCEP stock solution should have pH adjusted with 0.1 M NaOH to 6.0, in order to prevent protein precipitation from pH changes.
- After incubation, filter the reduced protein using 0.2 µm filter to remove protein precipitates.
- To the new reaction tube add first four-fold molar excess of MMAE (dissolve MMAE in N,N-dimethylacetamide (DMAc) to prepare 40 mM stock solution) over protein -SH groups. Then add double volume of PBS buffer in relation to the volume of the Fc protein. At last add reduced Fc fragment from step 1.2.
NOTE: In this step, order of reagents addition is important. - Carry out the conjugation reaction overnight at 15 ˚C with gentle rotation. These mild conditions prevent protein denaturation.
- Filter the solution with conjugate using 0.2 µm filter to remove potential precipitates.
- Carry out SDS-PAGE to analyze the efficiency of the reaction. Use 10% separation gel, 10 – 250 kDa protein marker, and analyze bands between 35 and 50 kDa (Figure 2).
NOTE: The protocol can be paused here, and the reaction mixture stored either at 4 ˚C for short term storage, or at -80 ˚C for long term storage. However, freezing and thawing can cause additional precipitation and decrease conjugate purification yields. - Load the reaction mixture on a column with protein A-conjugated beads and wash off the excess of MMAE with Washing buffer 1 containing 300 mM NaCl, 18 mM NaH2PO4, 33 mM, Na2HPO4, 0.1% Tween 20, 2 mM EDTA pH 7.4. Then wash the column using Washing Buffer 2 with 650 mM NaCl, 18 mM NaH2PO4, 33 mM Na2HPO4, pH 7.4.
NOTE: Through selective interaction of Fc region of the conjugate with protein A on the beads, the conjugate is captured on the column, while unreacted MMAE is washed out. This step is necessary to obtain highly pure conjugate. - Elute the Fc-MMAE conjugate from the column with 0.1 M sodium citrate pH 3.5 to 1.5 mL tubes containing 1 M Tris pH 9.0 (1 mL of protein to 200 µL of 1 M Tris pH 9.0).
- Desalt Fc-MMAE to PBS pH 7.4 using a column with Sephadex G-25 resin. This step prevents denaturation of the conjugate and allows the conjugate to be used in in vitro and in vivo tests.
NOTE: The protocol can be paused here. - Carry out SDS-PAGE (10%) to analyze the final Fc-MMAE conjugate. Use 10% separation gel, 10 – 250 kDa protein marker, and analyze bands between 35 and 50 kDa (Figure 2).
2. Conjugation of engineered FGF2
- Conjugation of GGGG-MTX to the C-terminal LPETGG sequence of engineered FGF2 via sortase A-mediated ligation
- Transfer purified engineered FGF2 containing N-terminal KCKSGG and C-terminal LPETGG sequence (used in this step for conjugation) to the sortase A Reaction Buffer (25 mM HEPES pH 7.4, 150 mM NaCl, 10 mM (NH4)2SO4, 5 mM CaCl2) using a column with G-25 resin. Adjust the protein concentration to 1 µM with sortase A Reaction Buffer.
- Add GGGG-MTX peptide dissolved in DMAc, directly to the protein solution to a final concentration of 100 µM.
- Add sortase A to a final concentration of 0.1 µM and incubate for 12 h at 15 °C with gentle rotation. The concentrations of reagents to be used in steps 2.1.1-2.1.3 are largely determined by the yield of conjugate to be achieved and the biochemical characteristics of the targeting protein (protein purification effectiveness, its solubility and stability). For effective sortase A reaction, use a starting point ten- to hundred-fold times lower concentration of sortase A than the LPETGG-containing target protein and 100 times higher concentration of GGGG-MTX than LPETGG-tagged protein.
- Load the reaction mixture on a Heparin Sepharose column.
- Wash off unreacted molecules with 25 mM HEPES pH 7.4.
- Elute the product of the reaction (FGF2.MTX) with 25 mM HEPES pH 7.4 with 2 M NaCl.
NOTE: The protocol can be paused here. - Analyze the efficiency of the conjugation using SDS-PAGE. Use 15% separation gel, 10 – 250 kDa protein marker, and analyze bands between 15 and 25 kDa (Figure 3).
- Conjugation of MMAE to the N-terminal KCKSGG sequence of engineered FGF2 via maleimide-thiol reaction
- Desalt engineered FGF2 containing N-terminal KCKSGG and C-terminal LPETGG sequence to the Reaction Buffer (25 mM HEPES pH 7.0, 10 mM (NH4)2SO4, 10 mM methionine, 0.1 mM EDTA) using a column with Sephadex G-25 resin. Adjust the protein concentration to 25 µM with the Reaction Buffer.
- Dissolve MMAE in DMAc to prepare 40 mM stock solution.
- To the new reaction tube add first four-fold molar excess of MMAE over protein -SH groups. Then add double volume of PBS buffer in relation to the volume of the FGF2 protein. At last add FGF2 protein.
NOTE: In this step order of reagents addition is important. - Carry out the reaction for 1 h at 20 °C with gentle rotation.
- Load the reaction mixture on a Carboxymethyl (CM)-Sepharose column and wash off the excess of the unconjugated cytotoxic agent with 25 mM HEPES pH 7.4. These mild conditions prevent protein denaturation.
- Elute the MMAE.FGF2 conjugate from the column with 25 mM HEPES pH 7.4 with 0.5 M NaCl.
NOTE: The protocol can be paused here. - Analyze the efficiency of the conjugation using SDS-PAGE. Use 15% separation gel, 10 – 250 kDa protein marker, and analyze bands between 15 and 25 kDa (Figure 3).
- Preparation of dual warhead conjugate of enginnered FGF2 using combination of maleimide-thiol chemistry and sortase A-mediated ligation
NOTE: For the generation of dual warhead conjugates of engineered FGF2 steps 2.1 and 2.2 are combined. First MMAE.FGF2 conjugate is produced via maleimide-thiol chemistry (step 2.2) and used for the attachment of GGGG-MTX with sortase A to the C terminal LPETGG tag of MMAE.FGF2 (step 2.1). Also, reverse procedure is also possible (first sortase A-mediated conjugation, then maleimide-thiol reaction).- Use steps 2.2.1-2.2.7 for the preparation of mono-substituted MMAE.FGF2 conjugate.
- Exchange the buffer for the sortase A Reaction Buffer (25 mM HEPES pH 7.4, 150 mM NaCl, 10 mM (NH4)2SO4, 5 mM CaCl2) using a column with Sephadex G-25 resin.
- Continue as described in 2.1.2-2.1.7.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
The presented protocols describe two distinct strategies for the conjugation of different cytotoxic drugs into proteins of interest. Furthermore, a combination of individual strategies is shown that allows to generate dual warhead cytotoxic conjugates in a site-specific manner.
As shown on SDS-PAGE gels in Figure 2 (lane 2 vs. 3) the maleimide-thiol reaction allows to reach almost 100% efficiency for the MMAE conjugation to the Fc fragment (visible as a shift of the Fc band due to the increased molecular weight resulting from MMAE incorporation). Since the Fc fragment has more than one putative conjugation site, we cannot fully exclude the possibility of mixture of mono- and dually-conjugated Fc in lane 3. To solve this point obtained conjugate can be further verified with mass spectrometry, or spectrophotometrically to determine drug to protein ratio, as described in Chen’s protocol20.
High efficiency of the conjugation is also obtained for engineered FGF2 variant using above-described protocols (Figure 3). The yield of sortase A-mediated ligation of MTX to the C-terminal LPETGG tag in the engineered FGF2 is close to 100% as estimated from SDS-PAGE (Figure 3, lane 3). Similarly, high efficiency of MMAE attachment to the N-terminal KCKSGG sequence of the engineered FGF2 is obtained via maleimide-thiol reaction (Figure 3, lane 4). When two reactions are combined, a dual warhead FGF2 conjugate (MMAE.FGF2.MTX) with MMAE and MTX attached in a site specific manner to the N- and C- termini of engineered FGF2, respectively is generated with almost 100% yield (Figure 3, lane 5).
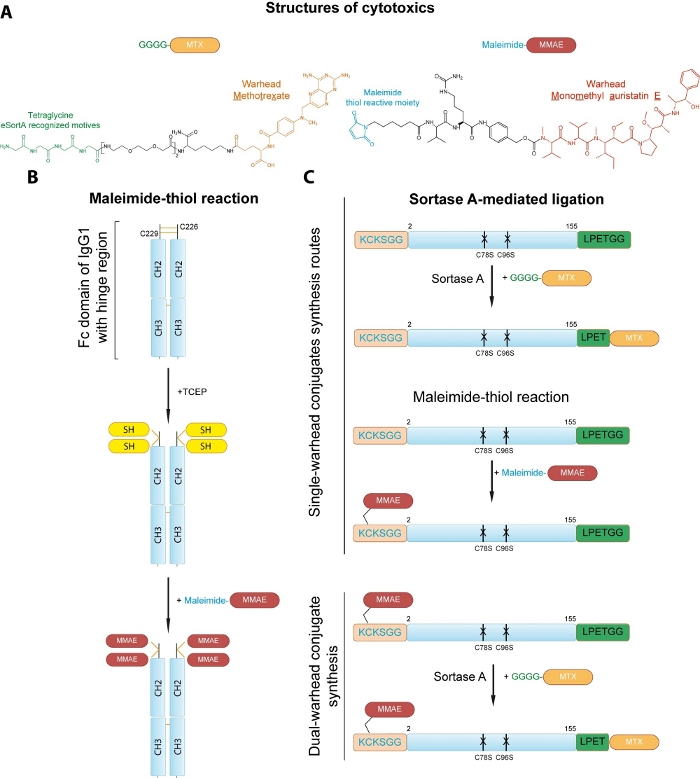
Figure 1: Strategies for site-specific conjugation of cytotoxic drugs to the proteins of interest. (A) Structures of drugs used for conjugation. (B) Conjugation of MMAE to the cysteine residues of the Fc fragment of human IgG1. Two disulfide linkages are initially reduced Cysteine residues are first reduced with TCEP to generate reactive thiol groups (SH) that are subsequently conjugated to the maleimide-MMAE. The dimeric assembly of the Fc is retained by the interaction between the constant heavy chain domains 2 and 3 (CH2 and CH3). (C) Development of single and dual warhead conjugate of engineered FGF2. Sortase A mediates attachment of the peptide-linked MTX to the C-terminal small LPETGG tag within FGF2 to yield FGF2.MTX. A maleimide-thiol reaction allows for the incorporation of the MMAE to the N-terminal KCKSGG sequence in engineered FGF2, resulting in MMAE.FGF2. Sortase A reaction allows for generation of MMAE.FGF2.MTX from MMAE.FGF2 and GGGG-MTX Please click here to view a larger version of this figure.

Figure 2: SDS-PAGE of the Fc fragment. SDS-PAGE of the Fc fragment before (lane 2) and after the conjugation with MMAE (lane 3). Lane 1 is a molecular weight marker. Please click here to view a larger version of this figure.
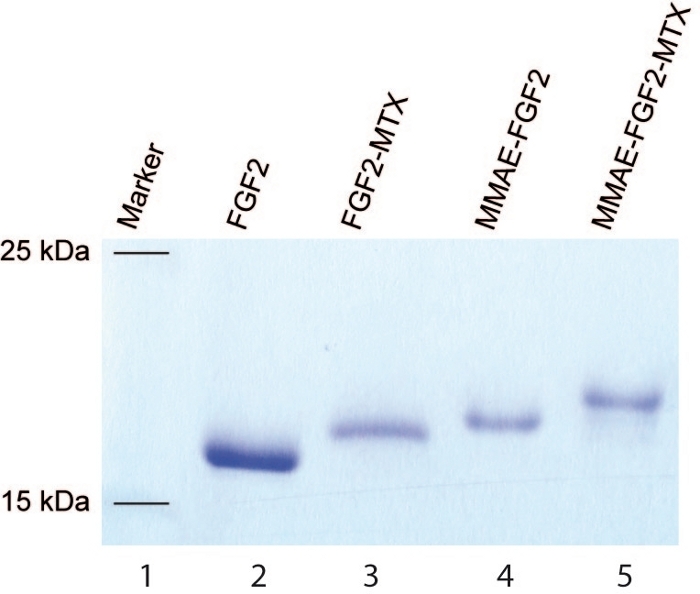
Figure 3: SDS-PAGE of the engineered FGF2. SDS-PAGE of the engineered FGF2 before (lane 2) and after the conjugation with MTX (lane 3), MMAE (lane 4) and MTX+MMAE (lane 5). Lane 1 is a molecular weight marker. Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Due to the high interest in the design of selective therapeutics against diverse cancer types there is an urgent need for strategies allowing for site-specific attachment of distinct cargoes to the targeting proteins. The site-specific modification of targeting proteins is critical as it ensures homogeneity of developed bioactive conjugates, a prerequisite for modern therapeutics. There are several methods, both chemical and enzymatic allowing for site-specific attachment of cargo to the protein of choice. In most cases these approaches require protein engineering steps prior to the generation of conjugate of interest. Depending on the selected method the preparation of protein might be laborious and expensive.
Here we provide a protocol in which two distinct strategies for site specific modification of proteins (one example of the Fc fragment of IgG1 and engineered FGF2) using chemical (maleimide-thiol chemistry) and enzymatic (protein ligation with sortase A) approaches are shown that highly efficient and relatively fast. The Fc fragment is a core component of therapeutic antibodies in the ADC approach, while engineered FGF2 is a modified growth factor that specifically targets FGFRs. Furthermore, we provide a protocol how two distinct strategies of conjugation can be combined to yield targeting protein with incorporated two distinct cargoes (in this case different cytotoxic drugs – MMAE and MTX).
A critical step in all presented approaches is the preparation of targeting protein. The protein should be highly pure (to ensure homogeneity of products), properly folded (to ensure biological activity) and often modified prior the conjugation (cysteine engineering, addition of small tags etc.). For homogeneity and site-specificity protein should be engineered to contain a single cysteine residue that is accessible for maleimide-thiol reaction. One possibility is to use naturally occurring surface-exposed cysteine within protein of interest with simultaneous mutation of other potentially reactive cysteines. Importantly, in contrast to coupling to the primary amines (α-amine group at the N-terminus of protein and the ϵ-amine group of lysine residues), conjugation to the cysteine residues does not change the net charge of a protein. If targeting protein lacks surface-exposed cysteine one can introduce this residue by site-directed mutagenesis, replacing amino acids non-essential for biological function (e.g., receptor binding), or for protein folding and stability. Exposed residues located in loops or unstructured regions should be preferentially used for cysteine substitution if the structural information is available for the modified protein. Alternatively, a small tag bearing cysteine flanked by basic residues for improved reactivity can be incorporated either at protein N or C termini. This approach was already employed with success for generation of cytotoxic FGF1 and FGF2 conjugates14,15.
Although approaches presented in these protocols require protein engineering steps, these modifications usually have no/little effect on targeting protein stability and biological activity. Furthermore, in contrast to laborious, expensive, and low-efficient incorporation of unnatural amino acids, these protein engineering steps still allow to achieve high yields of pure recombinant proteins.
The other critical step is the maleimide-thiol-based conjugation is efficient reduction of cysteine residues. In contrast, the sortase A-mediated ligation does not require modification of protein core, but it is strictly dependent on the availability of the C-terminal LPETGG peptide. Therefore, the C-terminal small tag should be incorporated into the protein of interest and if required, a linker between the protein core and the small tag should be introduced to ensure accessibility of sortase A to the LPETGG sequence. Moreover, sortase A-based method, in contrast to other ones (i.e., formylglycine-generating enzymes, transglutaminase, tubulin tyrosine ligase, trypsiligase, phosphopantetheinyl transferase or biotin ligase) is highly efficient, established amide bond is stable, reaction conducts in mild conditions, and do not require long or hydrophobic tags21.
Importantly, in some instances the conjugation can affect stability, structure, or function of the targeting protein. Therefore, it is critical to test the functionality of targeting protein in obtained conjugate (e.g., by analyzing receptor binding or receptor-mediated endocytosis) and its stability (e.g., by measuring the levels of conjugates in serum or cell-conditioned media in time)22,23,24.
The provided protocols were successfully used to develop conjugates with FGF1, FGF2 and antibody fragment-Fc as targeting molecules3,11,12,14,25,26,27. However, they can be, in principle, applied to any targeting factor of interest, including the Fc fragment-containing proteins like antibodies, antibody fragments or peptibodies. Presented methods can also be used in combination with other drugs to yield cytotoxic conjugates of proteins for anti-cancer therapies. Alternatively, these protocols can employ fluorophores to develop specific protein-based probes for the diagnostics.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the First TEAM and the Reintegration programs of the Foundation for Polish Science (POIR.04.04.00-00-43B2/17-00; POIR.04.04.00-00-5E53/18-00) co-financed by the European Union under the European Regional Development Fund, awarded to L.O and A.S. M.Z work was supported by OPUS (2018/31/B/NZ3/01656) and Sonata Bis (2015/18/E/NZ3/00501) from the National Science Centre. A.S.W work was supported by Miniatura grant from the National Science Centre (2019/03/X/NZ1/01439).
Materials
| Name | Company | Catalog Number | Comments |
| CM-Sepharose column | Sigma-Aldrich, Saint Louis, MO, USA | CCF100 | |
| Heparin Sepharose column | GE Healthcare, Chicago, IL, USA | GE17-0407-01 | |
| HiTrap Desalting column | GE Healthcare, Chicago, IL, USA | GE17-1408-01 | |
| HiTrap MabSelect SuRe column | GE Healthcare, Chicago, IL, USA | GE11-0034-93 | |
| maleimidocaproyl-Val-Cit-PABC-monomethyl auristatin E (MMAE) | MedChemExpress, Monmouth Junction, NJ, USA | HY-100374 | Toxic |
| N,N-Dimethylacetamide (DMAc) | Sigma-Aldrich, Saint Louis, MO, USA | 185884 | |
| Tris(2-carboxyethyl)phosphine (TCEP) | Sigma-Aldrich, Saint Louis, MO, USA | 646547 |
References
- Bethany, H. A new generation of antibody-drug conjugates for cancer patients. Chemical & Engineering News. 98 (14), (2020).
- Tu, C., et al. A Combination of structural and empirical analyses delineates the key contacts mediating stability and affinity increases in an optimized biotherapeutic single-chain Fv (scFv). Journal of Biological Chemistry. 291 (3), 1267-1276 (2016).
- Sokolowska-Wedzina, A., et al. High-affinity internalizing human scFv-Fc antibody for targeting FGFR1-overexpressing lung cancer. Molecular Cancer Research. 15 (8), 1040-1050 (2017).
- Seeger, M. A., et al. construction, and characterization of a second-generation DARPin library with reduced hydrophobicity. Protein Science. 22 (9), 1239-1257 (2013).
- Deyev, S., et al. Synthesis, characterization, and selective delivery of DARPin-gold nanoparticle conjugates to cancer cells. Bioconjugate Chemistry. 28 (10), 2569-2574 (2017).
- Currier, N. V., et al. Targeted drug delivery with an integrin-binding Knottin-Fc-MMAF conjugate produced by cell-free protein synthesis. Molecular Cancer Therapeutics. 15 (6), 1291-1300 (2016).
- Cox, N., Kintzing, J. R., Smith, M., Grant, G. A., Cochran, J. R. Integrin-targeting knottin peptide-drug conjugates are potent inhibitors of tumor cell proliferation. Angewandte Chemie International Edition. 55 (34), 9894-9897 (2016).
- Goldberg, S. D., et al. Engineering a targeted delivery platform using Centyrins. Protein Engineering Design and Selection. 29 (12), 563-572 (2016).
- Altai, M., et al. Affibody-derived drug conjugates: Potent cytotoxic molecules for treatment of HER2 over-expressing tumors. Journal of Controlled Release. 288, 84-95 (2018).
- Sochaj-Gregorczyk, A. M., Serwotka-Suszczak, A. M., Otlewski, J. A Novel affibody-Auristatin E conjugate with a potent and selective activity against HER2+ cell lines. Journal of Immunotherapy. 39 (6), 223-232 (2016).
- Szlachcic, A., Zakrzewska, M., Lobocki, M., Jakimowicz, P., Otlewski, J. Design and characteristics of cytotoxic fibroblast growth factor 1 conjugate for fibroblast growth factor receptor-targeted cancer therapy. Drug Design, Development and Therapy. 10, 2547-2560 (2016).
- Krzyscik, M. A., Opaliński, Ł, Otlewski, J. Novel method for preparation of site-specific, stoichiometric-controlled dual warhead conjugate of FGF2 via dimerization employing sortase A-mediated ligation. Molecular Pharmaceutics. 16 (8), 3588-3599 (2019).
- Hermanson, G. T. The Reactions of bioconjugation. Bioconjugate Techniques. , 229-258 (2013).
- Lobocki, M., et al. High-yield site-specific conjugation of fibroblast growth factor 1 with monomethylauristatin e via cysteine flanked by basic residues. Bioconjugate Chemistry. 28 (7), 1850-1858 (2017).
- Krzyscik, M. A., et al. Cytotoxic conjugates of fibroblast growth factor 2 (FGF2) with monomethyl auristatin e for effective killing of cells expressing FGF receptors. ACS Omega. 2 (7), 3792-3805 (2017).
- Chen, I., Dorr, B. M., Liu, D. R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proceedings of the National Academy of Sciences. 108 (28), 11399-11404 (2011).
- Weiner, E. M., Robson, S., Marohn, M., Clubb, R. T. The sortase A enzyme that attaches proteins to the cell wall of Bacillus anthracis contains an unusual active site architecture. Journal of Biological Chemistry. 285 (30), 23433-23443 (2010).
- Sokolowska-Wedzina, A., et al. Efficient production and purification of extracellular domain of human FGFR-Fc fusion proteins from Chinese hamster ovary cells. Protein Expression and Purification. 99, 50-57 (2014).
- Merrifield, R. B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. Journal of the American Chemical Society. 85 (14), 2149-2154 (1963).
- Chen, Y. Drug-to-antibody ratio (DAR) by UV/Vis spectroscopy. Methods in Molecular Biology. 1045, 267-273 (2013).
- Falck, G., Müller, K. M. Enzyme-based labeling strategies for antibody-drug conjugates and antibody mimetics. Antibodies. 7 (1), Basel, Switzerland. 4 (2018).
- Shental-Bechor, D., Arviv, O., Hagai, T., Levy, Y. Folding of conjugated proteins. Annual Reports in Computational Chemistry. , Elseiver. 263-277 (2010).
- Bigman, L. S., Levy, Y. Entropy-enthalpy compensation in conjugated proteins. Chemical Physics. 514, 95-105 (2018).
- Danielsson, J., et al. Thermodynamics of protein destabilization in live cells. Proceedings of the National Academy of Sciences. 112 (40), 12402-12407 (2015).
- Świderska, K. W., Szlachcic, A., Czyrek, A., Zakrzewska, M., Otlewski, J. Site-specific conjugation of fibroblast growth factor 2 (FGF2) based on incorporation of alkyne-reactive unnatural amino acid. Bioorganic & Medicinal Chemistry. 25 (14), 3685-3693 (2017).
- Krzyscik, M. A., Zakrzewska, M., Otlewski, J. Site-specific, stoichiometric-controlled, PEGylated conjugates of fibroblast growth factor 2 (FGF2) with hydrophilic auristatin Y for highly selective killing of cancer cells overproducing fibroblast growth factor receptor 1 (FGFR1). Molecular Pharmaceutics. 17 (7), 2734-2748 (2020).
- Borek, A., Sokolowska-Wedzina, A., Chodaczek, G., Otlewski, J. Generation of high-affinity, internalizing anti-FGFR2 single-chain variable antibody fragment fused with Fc for targeting gastrointestinal cancers. PLOS ONE. 13 (2), 0192194 (2018).