

Summary
土壌微生物群集から、高分子量、高品質のゲノムDNAを単離する方法論が説明されています。
Abstract
土壌のmicrobiomeは密接に陸域生態系内の栄養とエネルギーフローに関連付けられているゲノムの多様性と代謝革新の広大な、比較的未踏の貯水池です。また、メタゲノムとして知られている耕作に依存しない環境ゲノム、、アプローチは、価値の高い治療とバイオマスの変換プロセスのための経路の再構築と機能的スクリーニングに関してこの遺伝情報への前例のないアクセスをお約束します。しかし、土壌のmicrobiomeは、まだ大規模な挿入ライブラリの生産のための十分な品質の高分子量のDNAを得ることが困難が主な原因課題である。ここでは、土壌や堆積物から高分子、微生物群集のゲノムDNAを抽出するためのプロトコルを導入する。単離したゲノムDNAの品質は、下流のシーケンシングとスクリーニングのアプリケーションに挿入する大規模な環境ゲノムライブラリーを構築するために最適です。
手順は、細胞溶解から始まります。微生物の細胞壁と膜は機械的(研磨)と化学力(β-メルカプトエタノール)の両方によって溶解されています。ゲノムDNAを抽出用緩衝液、クロロホルム - イソアミルアルコールとイソプロピルアルコールを用いて単離されている。溶解および抽出工程に用いる緩衝液は、高分子量のゲノムDNAの完全性を維持するためにグアニジンイソチオシアネートと臭化ヘキサデシルトリメチルアンモニウム(CTAB)などがあります。ダウンストリームアプリケーションに応じて、単離されたゲノムDNAをさらに塩化セシウム(CsCl密度)フミン酸を含む不純物を減少させる勾配超遠心を用いて精製することができる。最初の手順で、抽出は、DNAの定量化のステップを除く、約8時間かかります。 CsCl密度勾配超遠心分離、二日間のプロセスです。全体の手順の実行中に、ゲノムDNAが剪断を防ぐために優しく扱われるべき、重大なボルテックス、および反復的な過酷なピペッティングを避ける。
Protocol
パートI:DNAの抽出
手順
1。 8時間は、DNAの定量を除く6つのサンプルを処理するために、必要とされています。
もし抽出を開始する前に、プレチル乳鉢、乳棒と-20℃の冷凍庫でへらまたは液体窒素を使用してする必要があります。また、氷上で20 mlのクロロホルムイソアミルアルコール(24:1)を含むプレチル50mlチューブ。 65にハイブリダイゼーションオーブンを設定° Cに予熱。それが℃の結晶を溶かすために60に暖かい結晶化されている場合、CTAB溶液を確認してください。ただ、抽出を開始する前に、最後の成分を添加することにより変性溶液と抽出緩衝液を完了します(下のレシピを参照)。ヒュームフード内のすべての抽出手順を行う。
2。変性緩衝液と抽出緩衝液を準備します。
注:DNA収量を最大にするために、それは正確に指示に従って、バッファを作成することが重要です。
3。あらかじめ冷却した乳鉢で土壌2gを置きます。
注:フィールドで採取した土壌は、輸送用のドライアイス上で凍結し、-80℃で保存してください。前のDNA抽出〜2 mmのふるいを使用して解凍し、ふるいの土壌。あなたは、最大10 gに土壌の開始量を増やすことができます
4。土壌へのソリューションを変性液1mlを追加。
5。土壌粒子は、粉末状と均質に見えるまで、乳棒を用いて2液Nの土壌を凍結して挽く。凍結と二回研削を繰り返します。
注意:完全に土壌試料をカバーする十分な液体窒素を用いて、研削中にフリーズ土壌のサンプルを保管してください。研削時のわずかな融解は許容されます。
6。あらかじめ冷却したスパチュラを用いて50 mlコニカルチューブにグラウンドの土を移す。
注:グランド土壌試料は-80℃この時点で、必要に応じてで保存できます。
7。抽出緩衝液9mlのを追加。ソリューションと土壌、低速で渦を簡単に混合する。
注:バッファが均質であることを確認するには、60℃に温め° Cを使用前に10-15分ほど。抽出全体で60 ° Cでバッファの残りをしてください。速度8でボルテックス(10が最大のスケールで)。
8。ハイブリダイゼーションオーブンで65℃で40分間インキュベートする。インキュベーションの間、連続して最低速度でチューブを回転させ、または軽くチューブを10分ごとに反転。
注:バッファが大きい表面積にわたって土壌に連絡できるように、ハイブリダイゼーションオーブンで水平にチューブを置きます。解決策は、インキュベーションの間に少し粘性が見えるかもしれません。
9。 14℃ - 10℃10分間、1800 × gで遠心する20ミリリットルのクロロホルム - イソアミルアルコール(24:1)を含む予備冷却50 mlコニカルチューブに上清を移す、氷上に置きます。
注:上清転送するとき、有機層(最下層)を妨害しないように注意してください。それはクリーンな上清を収集するために必要がある場合上清の1〜2 mlのままにします。あなたは多くの場合、上清の上にベージュ層(厚さは約1-3 mm)を見ることができます。上清を収集するときにこの層を邪魔しないでください。このベージュ層の破損を防ぐために、管壁に沿って先端をスライドしてみてください。上清はあまりに濁っている場合は、クロロホルム - イソアミルアルコール抽出上清の1つのより多くの時間を繰り返す。クロロホルムを転送する際にクロロホルムがプラスチックを溶かすので、使い捨てのプラスチック製のピペットを使用しないでください。ガラスピペットを使用して、またはガラスのメスシリンダー、または50 mlコニカルチューブに直接クロロホルムを注ぐ。
10。土壌のペレットを2回以上を抽出する。
10.1。土壌のペレットを5 mlの抽出緩衝液を加え、穏やか低速で簡単にボルテックスに続いて1 mlの先端を使って混ぜる。
注意:使用前にバッファを振って混ぜる。
10.2。 65℃のハイブリダイゼーションオーブンで10分間インキュベートする。 14℃ - 10℃10分間、1800 × gで遠心する
ステップ9でチューブに上清を移す。チューブ回収した上清とクロロホルム - イソアミルアルコールで抽出中に氷上にしてください。
注:サンプル間混合クロスすることなく、同じチューブに上清を追加することを確認してください。
10.3。 9.1と9.2つのより多くの時間を繰り返します。
11。回転プレートを使用して、非常に静かに採取した上清(14〜19ミリリットル)と10分は1.25 rpmでクロロホルム - イソアミルアルコールを含むチューブを揺らし。
注意:漏れを防ぐために非常にしっかりと蓋を閉じてください。の漏れを容易に検出できるように、チューブを配置する前にロッキングプレート上にペーパータオルを置きます。
12。 14℃ - 10で20分間1800 × gの遠心
13。新しいチューブに水相を移す。
注:水相(上部層)と有機層(底部との間の相間を邪魔しないでください。層)に、上清を移す。唯一のクリーンな上清のコレクションを確保するために上清約1.5 2mlを残す。
14。イソプロピルアルコールを( - 21.aステップ15.a)を使用してDNAを回収する。あなたが回復することは困難であるイソプロピルアルコール沈殿によりほとんど見えないペレットを生成するサンプル内のバイオマスの非常に低い金額を、予想される場合 - あるいは、アミコン®ウルトラ- 15遠心フィルターデバイス(21.bステップ15.b)を使用します。
15.a.新しいチューブの超遠心上清を置きます。
16.a. 0.6:1の容積比で上清を回収するためにイソプロピルアルコールを追加。ミックスに軽く数回チューブを反転。
室温で30分間17.aインキュベートする。
18.a. 20℃20分間16000 × gで遠心分離、 - 25℃遠心分離のステップの前によくバランスされているすべてのチューブの重みを確認してください。
注:チューブの遠心力の方向をマーク、そのDNAの収量が低いときに、より簡単にDNAペレットを検出することができます。
19.a.吸引デカントまたは吸引により、イソプロピルアルコールを削除します。 DNAペレットを乱さないように注意してください。
注意:できるだけ早く遠心分離が完了するとイソプロピルアルコールを削除します。 DNAペレットをなくさないように十分注意してください。 DNAペレットのサイズは(〜9 mmの直径)はっきり見えるからほとんど見えないため、土壌の種類に応じて大きく異なる場合があります。それは慎重になるようではないDNAペレットから吸引チューブの壁に沿って吸引によるイソプロピルアルコールの残留物を除去することをお勧めします。
20.a. 5室温で乾燥したDNAペレット - 20分。
注:乾燥したDNAペレット以上しないでください、それ以外の場合はDNAを再溶解が困難になる。
21.a. 200μlのTEにDNAペレットを再懸濁します。穏やかなタッピングで混ぜる。 1.5 mlチューブにDNA溶液を移す。 ℃で一晩完全にDNAを溶解させるために4でチューブを保持します。
注:ペレットのサイズに応じて、追加するTEの音量を調整することができます。通常、200 - TE 400μlのが適しています。
21.b. - *あるいは手順15.bに従ってください
15.b.アミコン®ウルトラ- 15遠心フィルターデバイスを事前に洗浄、10分間、3500 × gでデバイスと遠心に15ミリリットルTEを追加。フロースルーを捨てる。
16.b.ステップ13からプレ洗浄アミコンフィルターにDNA溶液を置きます。 DNA量が200に減少するまで3500 × gで遠心する- 500μlを。フロースルーを捨てる。
17.b.二回TEにDNAを洗浄する。アミコンフィルターデバイスに10ミリリットルTEを追加。 DNA量は約200μlに減少するまで3500 × gで遠心します。フロースルーを捨てる。複数の時間を繰り返す。
18.b.新しい1.5 mlチューブにフィルターにDNA溶液を移す。
19.b.アミコンフィルターに40μlのTEを追加。フィルター上の任意のDNAの残留物を回収するためにピペッティングすることにより、フィルタ膜の両方の側面を洗ってください。ステップ18.b.でチューブにこの洗浄液を追加
20.b.必要に応じて、DNAをさらにマイクロコン® Ultracel YM - 30遠心フィルターユニットにより濃縮することができます。200μlのTEを加えると7分には、10000 × gで遠心分離することにより、フィルタを事前に洗ってください。 1分間5000 × gでフィルターや遠心分離にDNA溶液を追加。フィルター上のDNA溶液の量は約50μlに減少するまで遠心操作を繰り返します。
注:マイクロコンフィルターの最大初期サンプル容量は500μlです。 14000 × gでオーバースピードで遠心分離していないメンブレンを完全に乾くまで遠心以上しないでください。いくつかの液体がまだ遠心分離後にフィルタをカバーしていることを確認してください。あなたが以上の遠心分離や膜が乾燥して、その後、15μlのTEを追加する場合、30秒間静かに撹拌し、21.b.に進みます
21.b. DNAを回収するために3分間1000 × gで新しいマイクロコンチューブと遠心分離機で逆さまにフィルターユニットを置きます。
22。 TAEゲル(0.8%、0.5xTAE、4時間50 V)によって、及び光度計分光光度計またはピコグリーンアッセイとプレートリーダーでDNAを定量化する。
注:DNAの量は、ゲル上に既知濃度(高分子DNAマーカーまたはλHindIII)とマーカーのバンドにサンプルのバンドを比較することによって推定することができます。
23。 (詳細については、ステップIIを参照して、少なくともその2 1%、1xTAEゲルでパルスフィールドゲル電気泳動(PFGE)を用いて高分子量のDNAの完全性チェックhttp://www.jove.com/index/details.stp?idを= 1387年 、土井:Taupp ら 10.3791/1387 2009)。。
注:代わりに低融点アガロースのこのQCの目的のために、通常のアガロースゲルを使用することができます。簡単に言うと、電気泳動条件は以下の通りである:2-250キロバイト、6ボルト、初期の切り替え時間:5秒、最終的なスイッチング時間:15秒、14で12時間実行℃にゲルでDNAを可視化するために1時間SYBRゴールド染色。
24。抽出したDNAの分子量が十分であれば、次のCsCl勾配遠心分離のステップに進みます。また、DNAをCsCl遠心する前に-20℃または-80℃で保存することができます。
DNAのメジアン径が同じであるか、大規模な挿入のメタゲノムDNAライブラリーを構築する場合、36 KBを超えている必要があります。
パートII。塩化セシウム密度勾配超遠心分離を用いてDNA精製
遠心分離機の調製:1.5 - 2時間
遠心分離のステップ:18時間
遠心分離後にエチジウムブロマイド(EtBrを)除去し濃縮したDNA:3時間
手順
詳細については、Wright らを参照してください。 、2009年http://www.jove.com/index/details.stp?id=1352 、DOI:10.3791/1352
代表的な結果/成果
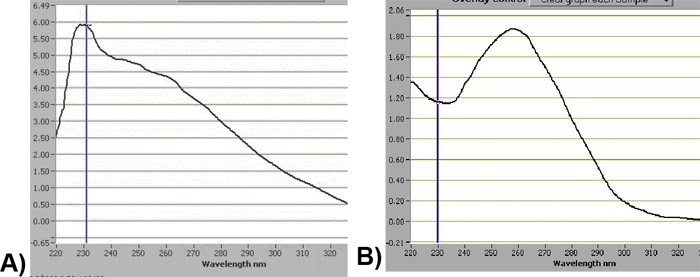
森林土壌有機物(O地平線)とミネラルの地平を2g(AE、AB、およびBtの地平線)から単離されたゲノムDNAの合計量(Skulow湖、BCにある長期の土壌の生産性(LTSP)のサイトからサンプリングされた、カナダと)森林との範囲のBC省によって管理さは10mgから180mgの範囲であった。 60キロバイト(図1) - 単離したDNAのメジアン径は40との間で普通であった。 260 nm/280 nmでの吸光度の比は1.3から1.6の間であったとしばしば土壌のサンプルで測定されたフミン酸の汚染を示す可能性のある230 nmにおけるピークがあった。 CsCl密度勾配遠心分離は、大幅にこの汚染を削減し、さらに280分の260〜1.8の割合の増加 - 図2a及び2bに示すように1.9。

塩化セシウム超遠心分離の前に抽出したゲノムDNAの図は、1。PFGE画像。 lane1:1キロバイトのマーカーは100 ng、レーン2:λHindIIIマーカーは100 ng、レーン3:λHindIIIマーカー250 ngの、レーン4:λHindIIIマーカー500ngの、lane5:フォスミド36キロバイトマーカーは100 ng、レーン6および7:ゲノムミネラル土壌地平線AE、レーン8および9から単離されたDNA:Skulow湖、BCのあるLTSPのサイトで鉱質土壌の地平線のABから単離したゲノムDNA

図2の前に()およびCsCl超遠心(B)の後に分光光度計によって検出されたゲノムDNAのChromograms。ゲノムDNAの質の改善を示す塩化セシウム超遠心法、後の230nmの波長での吸光度値の減少に注意してください。してくださいここをクリックして図2の拡大バージョンを参照すること。
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
我々は、イノベーションのためのカナダの財団、ブリティッシュコロンビア州ナレッジ開発基金、ゲノムブリティッシュコロンビア州と森林土壌の生産性の継続的な研究をサポートするための森林との範囲のブリティッシュコロンビア州省に感謝します。 SLは、微生物の多様性と進化のためのトゥーラ基礎資金センターからのフェローシップでサポートされていました。
Materials
| Name | Company | Catalog Number | Comments |
| *Denaturing solution: 10 ml in total | |||
| Guanidine isothiocyanate (MW 118.16), 4.73 g | |||
| 1M Tris-HCl (pH 7.0), 100 μl | |||
| 0.5M EDTA, 20 μl | |||
| Add water to 9.95 ml in total. | |||
| Autoclave. | |||
| Add 50 μl 2-mercapt–thanol just before use. (5 μl of 2-mercapt–thanol per 1 ml Denaturing Solution) | |||
| Note: Keep the Denaturing solution at 4 °C. Do not use buffer older than one week. If possible, make fresh buffer to use. | |||
| **Extraction Buffer | |||
| 1M Sodium phosphate buffer [pH 7.0]*, 100 ml | |||
| 1M Tris-HCl [pH 7.0], 100 ml | |||
| 0.5M EDTA [pH 8.0], 200 ml | |||
| 5 M NaCl, 300 ml | |||
| Autoclave and keep it at room temperature. | |||
| Add 200ml, 5% Hexadecyltrimethylammonium bromide (CTAB, autoclaved) and 100 ml, 20% SDS (autoclaved) just before use. If CTAB was crystallized, melt it at 60 °C. | |||
| * 1M Sodium phosphate buffer: 57.7 ml, 1M Sodium phosphate monobasic (NaH2PO4) and 42.3 ml, 1M Sodium phosphate dibasic (NaH2PO4). Adjust pH to 7.0 | |||
| Note: Do not use 20 % SDS if it has precipitation. It is normal to see milky suspension when you add SDS to the solution. Once you add SDS, place the extraction buffer at 60 °C to ensure SDS is well suspended. | |||
References
- Hurt, R. A., Qiu, X., Wu, L., Roh, Y., Palumbo, A. V., Tiedje, J. M., Zhou, J. Simultaneous recovery of RNA and DNA from soils and sediments. AEM. 67 (10), 4495-4503 (2001).
- Wright, J. J., Lee, S., Zaikova, E., Walsh, D. A., Hallam, S. J. DNA extraction from 0.22 μM Sterivex filters and cesium chloride density gradient centrifugation. JoVE. , (2009).
- Taupp, M., Lee, S., Hawley, A., Yang, J., Hallam, S. J. Large insert environmental genomic library production. J Vis Exp. , (2009).

{kind=link}