

Summary
इस लेख जीवाणुभोजी lambda, संक्रमण के एक fluorescently लेबल संस्करण की तैयारी के लिए प्रक्रिया का वर्णन
Protocol
1. एक कच्चे फेज lysate का निर्माण (1 चित्रा)
- एक 50 मिलीलीटर फ्लास्क में, LE392 के एक ताजा कॉलोनी (LZ1 λ) [pPLate * डी] (विवरण के लिए 1 टेबल देखें) लेग 7 मध्यम 10 / ग्राम मिलीलीटर केनामाइसिन और 100 छ / मिलीलीटर एम्पीसिलीन साथ पूरक 6 मिलीलीटर में टीका लगाना. 30 ° हल्के झटकों (180 rpm) के साथ सी रातोंरात आगे बढ़ें.
- LBM (10 मिमी MgSO 4 के साथ पूरक लेग) में संस्कृति 1:100 पतला और हल्के झटकों (180 rpm) के साथ 30 डिग्री सेल्सियस से बढ़ने की. आदेश में फेज उपज का अनुकूलन करने के लिए, यह सुनिश्चित करें कि संस्कृति की मात्रा अधिक नहीं फ्लास्क मात्रा क्षमता की एक 10 से अधिक है. हम आम तौर पर 2 लीटर या 2.5 लीटर क्षमता की दो बोतल तैयार है, और प्रत्येक फ्लास्क में 250 मिलीलीटर LBM मध्यम में 2.5 मिलीलीटर रातोंरात संस्कृति जोड़ने.
- जब सेल घनत्व 600 आयुध डिपो तक पहुँचता ≈ 0.6 (~ 2.5 - 3 घंटा), हल्के झटकों (180 rpm) के साथ 18 मिनट के लिए एक 42 डिग्री सेल्सियस पानी स्नान हिलनेवाला संस्कृति हिल द्वारा lysogen प्रेरित, और फिर incubat37 पर ई ° C lysis जब तक हल्के झटकों (180 आरपीएम) के साथ दिखाई देता है (संस्कृति स्पष्ट हो जाता है, 60 ~ 90 मिनट).
- संस्कृति 2% क्लोरोफॉर्म जोड़ें, मिश्रण करने के लिए हाथ से हिला, और फिर कमरे के तापमान पर 15 मिनट के लिए सेते हैं. चेतावनी: के लिए क्लोरोफॉर्म संभाल दस्ताने पहनें, और उसे साँस लेने से बचें.
- दो 250 मिलीलीटर अपकेंद्रित्र बोतलों में संस्कृति स्थानांतरण, एक Sorvall जीएसए रोटर में 4 डिग्री सेल्सियस पर 10,000 rpm पर संस्कृति अपकेंद्रित्र 15 मिनट के लिए पुनर्प्राप्त फेज कणों से युक्त तैरनेवाला, और मलबे की गोली त्यागें. प्रदर्शन दिखाई मलबे से छुटकारा पाने के लिए सुनिश्चित करने के लिए एक दूसरे centrifugation.
- फेज एकाग्रता को मापने के लिए एक मानक फेज अनुमापन 8 प्रोटोकॉल का उपयोग करने के लिए. फेज अनुमापांक होना चाहिए ~ 5-10 10 9 एक्स / pfu मिलीलीटर. LE392 फ्लोरोसेंट फेज के जीनोटाइप में SAM7 उत्परिवर्तन के कारण सूचक तनाव के रूप में के रूप में एक supF तनाव का प्रयोग करें, और शीर्ष अगर और अगर प्लेटों के अमीर NZYM साथ बनाया बड़ा सजीले टुकड़े प्राप्त करने का उपयोग करें (फाईgure 2).
2. फेज शोधन (1 चित्रा)
- एक बड़े कुप्पी (उदाहरण के 2 लीटर) में डालो lysate, lysate DNase मैं और RNase (1 ग्राम / मिलीलीटर प्रत्येक) जोड़ने के क्रम में न्यूक्लिक lysed बैक्टीरिया से मुक्त एसिड को पचाने और कमरे के तापमान पर 1 घंटा सेते हैं.
- Lysate 1M NaCl जोड़ें, 250 मिलीलीटर अपकेंद्रित्र बोतलों में lysate हस्तांतरण, और बर्फ पर 3 घंटा सेते हैं. 4 पर 15 मिनट के लिए 10,000 rpm पर एक Sorvall जीएसए में lysate अपकेंद्रित्र डिग्री सेल्सियस तैरनेवाला पुनर्प्राप्त. फेज अनुमापांक कच्चे lysate, जो है कि समान होना चाहिए ~ 5-10 10 9 एक्स / pfu मिलीलीटर. NaCl के अलावा बैक्टीरियल मलबे से फेज कणों की हदबंदी को बढ़ावा देता है और 8 खूंटी द्वारा फेज कणों के कुशल वर्षा के लिए आवश्यक है.
- एक बड़े फ्लास्क में डालो lysate, जैसे कुप्पी 2 लीटर, lysate में 10% (w / v) PEG8000 जोड़ने, धीरे धीरे हलचल या कमरे के तापमान पर PEG8000 भंग हिला. 250 मिलीलीटर प्रतिशत में lysate स्थानांतरणrifuge बोतलें और फिर रातोंरात (~ 16 घंटा) 4 डिग्री सेल्सियस पर सेते हैं 4 पर 15 मिनट के लिए 10,000 rpm पर एक Sorvall जीएसए रोटर में lysate अपकेंद्रित्र डिग्री सेल्सियस तैरनेवाला त्यागें.
- फेज एस.एम. बफर (प्रारंभिक फेज lysate की 250 मिलीलीटर प्रति 4 मिलीलीटर एस.एम. बफर) के साथ गोली (फेज कणों PEG8000 साथ उपजी) भिगोएँ. बहुत मिलाते हुए हल्के या नहीं 4 डिग्री सेल्सियस पर 16 घंटे के लिए झटकों के साथ सेते
- धीरे lysate (फेज कणों के साथ एस.एम. बफर) एक 50 मिलीलीटर Eppendorf अपकेंद्रित्र ट्यूब में लेने के लिए, और फिर 0.5 के साथ शेष गोली धोने एस.एम. बफर के 1 मिलीलीटर.
- Lysate क्लोरोफॉर्म की बराबर मात्रा में जोड़ें. धीरे क्लोरोफॉर्म साथ inverting ऊपर और नीचे से कुछ समय के लिए lysate मिश्रण. एक Eppendorf 5804R में 15 डिग्री सेल्सियस 4 मिनट या एक समान पीठ टॉप अपकेंद्रित्र के लिए 4000 rpm पर अपकेंद्रित्र.
- एक स्पष्ट lysate 2.6 चरण दोहराएँ. फेज अनुमापांक ~ 1-2 होना चाहिए x 10 11 / pfu मिली.
- 1.3 छ / मिलीलीटर, 1 के तीन अलग घनत्व (ρ) के साथ एस.एम. / CsCl समाधान तैयार.5 ग्राम / मिलीग्राम और 1.7 ग्राम / एमएल. अपवर्तक सूचकांक (η) को मापने के लिए एक और अधिक सटीक पढ़ पाने के घनत्व. घनत्व 9 रूपांतरण ρ = η 10.8601-13.4974 पर 25 डिग्री सेल्सियस विवरण के लिए 3 टेबल देखें.
- एक लंबी सुई के साथ एक सिरिंज का प्रयोग करने के लिए एक 14 मिलीलीटर अल्ट्रा स्पष्ट Beckman 40Ti ultracentrifuge ट्यूब में समाधान लोड. मिश्रण से बचने के लिए एक बेहतर घनत्व ढाल के रूप में करने के लिए, समाधान (यानी layering एक के तहत बढ़ती घनत्व के समाधान एक और) किया जाना चाहिए, यानी, 1.3 ग्राम / मिलीग्राम, 1.5 के क्रम में धीरे / एस.एम. CsCl समाधान के 2 मिलीलीटर लोड underlaying ग्राम / मिलीग्राम और 1.7 ट्यूब के नीचे करने के लिए एक 3 मिलीलीटर सिरिंज के साथ सुई डालने से छ / मिलीलीटर.
- धीरे 14 मिलीलीटर ultracentrifuge ट्यूब के ऊपर से overlaying द्वारा फेज lysate 8 मिलीग्राम लोड. एक संतुलन ट्यूब तैयार. 4 में 4 घंटे के लिए 24,000 rpm पर एक Beckman SW40Ti रोटर में अपकेंद्रित्र डिग्री सेल्सियस
- धीरे एक अंधेरे कमरे में ट्यूब बाहर ले और एक काले रंग की पृष्ठभूमि के खिलाफ ट्यूब के ऊपर से रोशन वायुसेना का उपयोग करlashlight. फेज बैंड 1.3 छ / एमएल और 1.5 ग्राम / मिलीग्राम / एस.एम. CsCl परतों (चित्रा 3 ए) के बीच इंटरफेस के स्थान पर स्पष्ट रूप से दिखाई जानी चाहिए. एक एक 3 मिलीग्राम सिरिंज के साथ 21.5 गेज सुई के साथ बैंड से थोड़ा नीचे ट्यूब के पक्ष के माध्यम से पंचर. धीरे से ~ फेज निलंबन के 500 μl इकट्ठा. फेज अनुमापांक ~ 5-10 10 11 x pfu / एमएल होना चाहिए.
- 4 मिलीलीटर अति स्पष्ट Beckman ultracentrifuge SW60Ti रोटर ट्यूब में फेज निलंबन रखें. 1.5 छ / एमएल एस.एम. / CsCl समाधान के साथ ट्यूब भरें. एक संतुलन ट्यूब तैयार. 4 में 24 घंटे के लिए 35,000 rpm पर एक Beckman SW60Ti रोटर में अपकेंद्रित्र डिग्री सेल्सियस
- 2.11 चरण में के रूप में एक ही प्रक्रिया दोहराएँ दिखाई बैंड से फेज इकट्ठा. बैंड के रूप में दिखाई हो चित्रा 3B में दिखाया जाना चाहिए.
- एक डायलिसिस झिल्ली कैसेट (2 तालिका) में फेज निलंबन लोड और 4 में एक 1000 गुना एस.एम. बफर की मात्रा डिग्री सेल्सियस के खिलाफ 3 घंटा, 3 घंटा और overnig durations के लिए तीन बार dialyzeht (~ 16 घंटा). डायलिसिस के प्रयोजन के लिए फेज निलंबन में मौजूद CsCl से छुटकारा मिल रहा है. अंतिम फेज अनुमापांक ~ 5-10 10 x 11 pfu / एमएल होना चाहिए.
3. एक agarose जेल स्लैब (4 चित्रा) तैयार
- 70% इथेनॉल के साथ 6 खुर्दबीन स्लाइड (75 x 50 मिमी, 1 मिमी मोटी) साफ करें.
- 5 स्लाइड्स व्यवस्थित और टेप के साथ सुरक्षित के रूप में 4 चित्र में दिखाया गया है.
- 6 लपेटो चिपटना (1.5% agarose उपज) के साथ एक छोटे से कवर बीकर में मिली माध्यम में 0.09 छ agarose मिक्स. एक गर्म थाली पर गर्मी जब तक समाधान स्पष्ट हो जाता है.
- सुरक्षित स्लाइड पर agarose समाधान डालो.
- शीर्ष पर पिछले स्लाइड प्लेस, ध्यान से हवाई बुलबुले से बचने. शीर्ष पर प्लेस वजन और ~ 30 मिनट के लिए शांत करने के लिए अनुमति देते हैं.
- पक्ष पर 4 स्लाइड निकालें और स्लैब लपेटो चिपटना के साथ ऊपर और नीचे स्लाइड के साथ साथ लपेटो. स्लैब 3 दिन के लिए 4 डिग्री सेल्सियस पर संग्रहीत किया जा सकता है.
4. शुद्ध फेज स्टॉक परीक्षण
- तैयार करनाएक जेल पीबीएस agarose स्लैब रूप में ऊपर वर्णित (धारा 3).
- DAPI साथ शुद्ध फेज दाग. मिक्स फेज की 10 μl (1 ~ 10 x 10 pfu / एमएल) 10 ग्राम / एमएल DAPI (5 ग्राम / एमएल की अंतिम DAPI एकाग्रता), 4 बजे 30 मिनट के लिए सेते हैं की 10 μl के साथ डिग्री सेल्सियस या कमरे के तापमान पर 10 मिनट.
- जगह एक 24 x 50 मिमी coverslip, उपरिशायी पूर्व तैयार स्लैब पीबीएस agarose का एक छोटा सा टुकड़ा नंबर 1 (~ 10 x 10 मिमी) के केंद्र में मिश्रण / फेज DAPI 1 μl. agarose स्लैब का छोटा सा टुकड़ा सैंडविच जेल निकाल दिया जाता है पर शीर्ष स्लाइड के बाद एक रेजर ब्लेड के साथ कट जाता है. YFP और DAPI चैनलों के माध्यम से epifluorescence खुर्दबीन के नीचे नमूना छवि. व्यक्तिगत phages विवर्तन सीमित दोनों चैनलों में फ्लोरोसेंट "स्पॉट '(चित्रा 5) के रूप में दिखाई जानी चाहिए. और नीचे 6.2 चरण में के रूप में एक ही खुर्दबीन कैमरा setups का उपयोग करें.
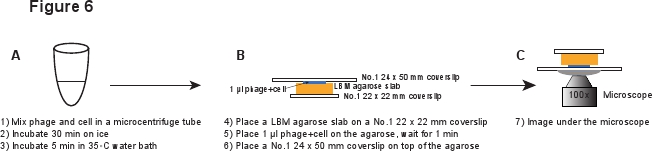
5. संक्रमण (6 चित्रा)
- एक 14 मिलीलीटर बाज़ ट्यूब में LE392 के एक ताजा कॉलोनी टीका लगाना [RE - मातृ एवं शिशु स्वास्थ्य पीपी] ERRY (विवरण के लिए 1 टेबल देखें) लेग 100 छ / मिलीलीटर एम्पीसिलीन, 10 मिमी MgSO 4 और 0.2% Maltose साथ पूरक मध्यम के 2 मिलीलीटर में. 37 डिग्री सेल्सियस पर मध्यम झटकों (265 rpm) के साथ रात में आगे बढ़ें.
- द्वारा LBMM (10 मिमी MgSO 4 और 0.2% Maltose साथ पूरक लेग) में संस्कृति 1:1000 पतला, यानी 5 मिलीलीटर LBMM मध्यम एक 50 मिलीलीटर फ्लास्क में में 5 μl रातोंरात संस्कृति जोड़ने. 37 ° मध्यम झटकों (265 rpm) के साथ सी 600 आयुध डिपो ≈ 0.4 आगे बढ़ें.
- LBM के माध्यम का उपयोग करने के लिए एक जेल LBM agarose स्लैब तैयार के रूप में ऊपर धारा 3 में वर्णित है.
- कमरे के तापमान पर 2 मिनट के लिए एक बेंच टॉप microcentrifuge में 2,000 ग्राम कोशिकाओं के 1 मिलीलीटर अपकेंद्रित्र. तैरनेवाला निकालें, और धीरे से 20 μl ठंडा करने के लिए 600 और 20 आयुध डिपो तक पहुँचने LBMM में कोशिकाओं resuspend.
- जब शुद्ध फेज स्टॉक से छेड़छाड़, एक विस्तृत विंदुक टिप का उपयोग करें या में कटौती करने के लिए नियमित रूप से विंदुक टिप टिप व्यापक खोलने, फेज 3 कणों कर्तन से बचने के. धीरे एमix फेज शुद्ध 0.1 की सीमा में एक औसत अनुपात phages सेल तक पहुँचने के 20 μl के साथ कोशिकाओं के 20 μl - 5. 30 मिनट के लिए बर्फ पर सेते फेज सोखना की अनुमति, और फिर एक 35 ° C नहाने के पानी में 5 मिनट के लिए सेते हैं फेज डीएनए इंजेक्शन 6 ट्रिगर.
- पिपेट और नीचे कुछ करने के लिए किसी भी सेल समुच्चय अलग बार. फिर एक विस्तृत विंदुक टिप का उपयोग phages कर्तन से बचने के लिए. मिश्रण 1:10 में LBMM, जैसे 5 45 μl LBMM में μl मिश्रण पतला.
- एक नंबर 1 22 x 22 मिमी coverslip पर LBM agarose स्लैब (~ 10 x 10 मिमी) का एक टुकड़ा रखें. पूर्व तैयार स्लैब LBM agarose कम से कम 1 घंटा के लिए कमरे के तापमान पर रखा जाना चाहिए का उपयोग करने से पहले यह सुनिश्चित करने के लिए कि agarose स्लैब कमरे के तापमान तक पहुँचता है. जगह agarose स्लैब पर मिश्रण / फेज सेल की 1 μl और 1 मिनट के लिए प्रतीक्षा करने के लिए मिश्रण agarose स्लैब में अवशोषित करने की अनुमति. धीरे agarose स्लैब के शीर्ष पर एक नंबर 1 24 x 50 मिमी coverslip जगह है. इस प्रक्रिया के लिए inf से phages कर्तन से बचने का मतलब हैected सेल (6 चित्रा).
6. खुर्दबीन के नीचे सेल भाग्य के बाद
- माइक्रोस्कोप के मंच पर ध्यान coverslip माउंट. इमेजिंग के लिए, एक उच्च बढ़ाई उद्देश्य (100x उदा) (नीचे चर्चा में माइक्रोस्कोप प्रणाली) का उपयोग करें.
- एक प्रारंभिक समय सीमा के लिए सेट छवि मोल. इस छवि को सेट करने के लिए प्रारंभिक सभी को संक्रमित करने phages की संख्या और पदों को चिह्नित करने के लिए इस्तेमाल किया जाएगा. 15 छवियों के 200 एनएम z-अक्ष (ऊर्ध्वाधर) अंतराल में एक श्रृंखला ले लो. YFP चैनल के माध्यम से छवि. इसके अलावा, चरण विपरीत और mCherry चैनलों के माध्यम से एक छवि में फोकस ले. प्रकाश की तीव्रता और समय जोखिम का अनुकूलन करने के लिए पर्याप्त संकेत प्राप्त करते हुए विरंजन और कोशिका क्षति को कम से कम (नीचे चर्चा में छवि अधिग्रहण).
- समय व्यतीत हो जाने के बाद संक्रमण सेल भाग्य की एक फिल्म मोल. चरण विपरीत YFP और mCherry में नमूना छविलगभग 4 घंटे के लिए 10 मिनट का समय अंतराल पर चैनल. फिल्म में समय व्यतीत हो जाने के दौरान, समय बिंदु प्रति चैनल के प्रति एक एकल छवि z-स्थिति का उपयोग करने के लिए, नमूना के अनावश्यक जोखिम से बचने के लिए, जो सफेद और phototoxicity नेतृत्व सकता.
7. छवि विश्लेषण
- मैन्युअल phages और रिकॉर्ड फेज और प्रारंभिक समय सीमा में सेल लंबाई स्थान की संख्या गिनती. इस Metamorph या ImageJ के रूप में इस तरह के सॉफ्टवेयर का उपयोग किया जा सकता है. सेल भाग्य (lytic, lysogenic या असंक्रमित), lysis समय, और फिल्म समय चूक खेल के द्वारा किसी भी अन्य वांछित जानकारी रिकार्ड. अलग सेल भाग्य की पहचान करने के लिए, प्रतिनिधि परिणाम के नीचे अनुभाग में समय चूक मूवी देखते हैं.
- मैनुअल उपरोक्त विश्लेषण के अलावा, और मात्रात्मक जानकारी (उदाहरण के लिए अलग - अलग कक्षों में समय पर प्रतिदीप्ति स्तर) स्वचालित सेल मान्यता और वंशावली अनुरेखण एल्गोरिदम का उपयोग करके निकाला जा सकता है. हम tr के लिए एक घर का निर्माण Matlab प्रोग्राम का उपयोग करेंसेल वंश और प्रतिदीप्ति स्तर acing, कोशिका विभाजन के के लिए Schnitzcell Matlab कोड (कैलटेक में Elowitz समूह द्वारा लिखित) के साथ एक साथ.
8. प्रतिनिधि परिणाम:
फेज चढ़ाना:
fluorescently लेबल phages (चरण 1.6 और धारा 2 में) के सजीले टुकड़े काफी जंगली प्रकार (2 चित्रा) के उन लोगों की तुलना में छोटे हैं. इसलिए हम प्लेटें 37 में कम से कम 12 घंटा सेते हैं सजीले टुकड़े के लिए डिग्री सेल्सियस इनक्यूबेटर दिखाई दे सकता है.
फेज ultracentrifugation:
CsCl कदम ढाल (2.10 चरण) के साथ फेज नमूना ultracentrifugation के बाद, दो बैंड दिखाई (चित्रा 3 ए) चाहिए. शीर्ष बैंड, फेज निलंबन और एस.एम. / CsCl 1.3 छ / मिलीलीटर परत के बीच इंटरफेस में, सेल मलबे और खाली फेज capsids शामिल हैं. नीचे बैंड, एस.एम. / CsCl 1.3 ग्राम / मिलीग्राम और 1.5 छ / मिलीलीटर परतों के बीच इंटरफेस में, फेज बैंड है. थीबैंड हरे फ्लोरोसेंट फेज λ LZ2 के लिए प्रकट होता है. जंगली प्रकार के के फेज λ IG2903 के लिए बैंड 5 नीले प्रकट होता है. 2.12 चरण में CsCl संतुलन ढाल ultracentrifugation के बाद, एक फेज बैंड ट्यूब (3B चित्रा) के मध्य भाग में दिखाई जानी चाहिए. चूंकि फ्लोरोसेंट फेज λ LZ2 GPD EYFP और GPD capsids, प्रोटीन डीएनए का अनुपात के एक मिश्रण होता है जंगली प्रकार की तुलना में अधिक है. इसलिए, फ्लोरोसेंट फेज λ LZ2 के बैंड थोड़ा कि जंगली प्रकार λ IG290310 की तुलना में हल्का (ट्यूब में एक उच्च स्थान पर प्रतीत होता है) है.
DAPI धुंधला:
चित्रा 5 ठेठ DAPI (धारा 4) के साथ फेज लेबल के बाद प्राप्त चित्रों से पता चलता है. YFP और DAPI संकेतों एक सफलतापूर्वक शुद्ध फेज के 100% के करीब पत्राचार होना चाहिए. हम आम तौर पर पालन कि YFP हाजिर के कम से कम 1%एस DAPI (वायरल जीनोम बिना capsids का प्रतिनिधित्व) शामिल नहीं है. DAPI स्पॉट की 1% से कम YFP (इसी गैर फ्लोरोसेंट phages) 5 शामिल नहीं है.
समय चूक मूवी:
Lytic कोशिकाओं YFP सेल के अंदर प्रतिदीप्ति (ग्रीन चैनल), सेल के संचय के द्वारा मान्यता प्राप्त हैं. Lysogenic कोशिकाओं सेल और सामान्य कोशिकाओं के विकास और विभाजन की बहाली के अंदर वर्दी mCherry प्रतिदीप्ति (लाल) के संचय के द्वारा मान्यता प्राप्त हैं. असंक्रमित कोशिकाओं (या कोशिकाओं जहां संक्रमण में नाकाम रही है) किसी भी ऊपर phenotypes नहीं प्रदर्शित करने के लिए और बढ़ने और सामान्य रूप से बांट देंगे. 7 चित्रा सेट चरण विपरीत कुछ छवि से पता चलता है, YFP और mCherry चैनल, और इनमें से इसी मढ़ा चित्र एक ठेठ फिल्म समय चूक (6 धारा) से तीन चैनलों. व्यक्तिगत phages (हरी स्पॉट) प्रारंभिक समय सीमा (चित्रा 7A) में स्पष्ट रूप से दिखाई दे रहे हैं. आमतौर पर, एक नंबरके phages कोशिका की सतह (संभवतः उन कोशिकाओं को संक्रमित) पर देखा जाता है जबकि अन्य phages unadsorbed कर रहे हैं, के रूप में चित्रा 7B (बाएं पैनल) में दिखाया गया है. संक्रमण के परिणाम समय पर अलग पहचाना हो जाता है. lytic चक्र नई phages के intracellular उत्पादन (हरे, 7C चित्रा) सेल (जारी हरी phages साथ विस्फोट कोशिकाओं, चित्रा 7D) द्वारा ने संकेत दिया है. Lysogeny पी रे (लाल, 7C चित्रा) प्रमोटर और सेल के विकास और विभाजन (लाल, चित्रा 7D) की बहाली से mCherry के उत्पादन ने संकेत दिया है.




चित्रा 1. एबी) को शरण देने के द्वारा प्राप्त की है. फेज कदम (पैनल सीएल) की एक श्रृंखला के माध्यम से शुद्ध होता है.

चित्रा 2 फेज सजीले टुकड़े. फ्लोरोसेंट फेज के सजीले टुकड़े (बाएं) जंगली प्रकार (दाएं) के उन लोगों की तुलना में 37 में 12 घंटे के लिए प्लेटें incubating के बाद छोटे ° सी.

चित्रा 3 ultracentrifugation बाद फेज बैंड.) दो बैंड CsCl कदम ढाल ultracentrifugation के बाद दिखाई दे रहे हैं. ऊपर एक सेल मलबे और खाली फेज capsids से मेल खाती है, नीचे बैंड वांछित फेज शामिल हैं. वाम: फ्लोरोसेंट फेज, सही: जंगली प्रकार बी एक एकल फेज बैंड) CsCl संतुलन ढाल ultracentrifugation के बाद दिखाई देता है. फ्लोरोसेंट फेज बैंड (बाएं) हरा है जंगली प्रकार फेज (दाएं) के लिए एक नीले बैंड की तुलना में.

चित्रा 4. Agarose जेल slabs की तैयारी की प्रक्रिया.

चित्रा 5 DAPI धुंधला हो जाना के बाद phages फ्लोरोसेंट छवियों. व्यक्तिगत phages आसानी से अलग पहचाना, और YFP हैं और DAPI का संकेत सह स्थानीय बनाना बहुत अच्छी तरह से.

चित्रा 6 फेज संक्रमण और इमेजिंग सेटअप के योजनाबद्ध वर्णन. इस छवि का एक पूर्ण आकार संस्करण देखने के लिए यहां क्लिक करें.
7 gure "" = "files/ftp_upload/3363/3363fig7.jpg /" />
7 चित्रा फेज संक्रमण के एक समय व्यतीत हो फिल्म से विशिष्ट छवियों. चरण विपरीत दिखाई हैं, YFP और mCherry चैनल, के रूप में के रूप में अच्छी तरह से तीन चैनलों के एक उपरिशायी. (ए) के प्रारंभिक समय सीमा से YFP चैनल छवियों. वाम, अलग z-पदों पर YFP छवियों के योग है. तीन सही छवियों नमूना अलग z-पदों, कोशिका की सतह के विभिन्न क्षेत्रों के लिए इसी पर YFP छवियों हैं. (बी), (सी) और (डी) (मध्य बाएं) चरण विपरीत, YFP (मध्यम दाएं) और mCherry अलग समय फ्रेम पर चैनल (दाएं) के मढ़ा चित्र (बाएं). (बी) टी = 0, दो कक्षों में देखा जाता है, एक सिंगल फेज (हरी स्पॉट), और एक सेल से संक्रमित प्रत्येक 3 phages से संक्रमित है. इसके अलावा मनाया कुछ unadsorbed phages कर रहे हैं. (सी) टी = 80 मिनट दो एकल phages से संक्रमित कोशिकाओं प्रत्येक lytic मार्ग में चले गए हैं, के रूप में संकेत मिलता हैनई phages के intracellular उत्पादन (हरा) घ. 3 phages द्वारा संक्रमित कोशिका lysogenic मार्ग, mCherry के पूर्व प्रमोटर (लाल) से उत्पादन से संकेत में चला गया है. (D) टी = 2 घंटा कम lytic मार्ग सेल (सेल विस्फोट) में है परिणामस्वरूप, जबकि lysogenic सेल § विभाजित किया गया है.
§ 7 चित्रा के वाम पैनल (सी) और (डी) सेल, 141 से reprinted हैं, Zeng, शमूएल स्किनर ओ, Chenghang Zong, जीन सिप्पी, माइकल Feiss, और Ido गोल्डिंग Lanying निर्णय एक Subcellular स्तर पर बनाने परिणाम निर्धारित करता है जीवाणुभोजी संक्रमण के 682-691, (2010) Elsevier से अनुमति के साथ कॉपीराइट.
| नाम तनाव | प्रासंगिक जीनोटाइप | स्रोत / संदर्भ |
| जीवाणु उपभेदों | ||
| LE392 | सुड़कनाएफ | जॉन Cronan, इलिनोइस विश्वविद्यालय |
| फेज उपभेदों | ||
| λ LZ1 | GPD EYFP, cI857 SAM7 D-eyfp ख :: kanR | एट अल 5. Zeng |
| λ LZ2 | GPD मोज़ेक, λ LZ1 के रूप में एक ही जीनोटाइप | एट अल 5. Zeng |
| प्लास्मिड | ||
| पीपी RE - mCherry | mCherry पी RE के नियंत्रण के तहत, amp आर | एट अल 5. Zeng |
| pPLate * डी | λ देर प्रमोटर के नियंत्रण के तहत GPD, amp आर | एट अल 5. Zeng |
तालिका 1 जीवाणु उपभेदों.phages और plasmids इस काम में इस्तेमाल किया.
| घनत्व ρ (छ / एमएल) | CsCl (छ) | एस.एम. (एमएल) | अपवर्तक सूचकांक η |
| 1.30 | 39 | 86 | 1.3625 |
| 1.50 | 67 | 82 | 1.3815 |
| 1.70 | 95 | 75 | 1.3990 |
तालिका 3. CsCl समाधान कदम gradients के लिए SM बफर (100 मिलीलीटर) में तैयार किया है.
Discussion
जीवाणु उपभेदों, फेज और प्लास्मिड:
LE392 तनाव supF है. यह फेज जीनोम में SAM7 उत्परिवर्तन को दबाने (विवरण के लिए 1 टेबल देखें) के लिए चुना गया था. इस प्रकार, प्रेरित lysogens अंततः lyse और फेज कणों रिलीज होगा, के रूप में संक्रमित कोशिकाओं कि lytic मार्ग चुना है. Lysogenic कोशिकाओं में बड़े हो रहे हैं 30 ° C CI फेज जीनोम में 857 एलील संवेदनशील तापमान की उपस्थिति के कारण. गर्मी प्रेरण के बाद, GPD EYFP और जंगली प्रकार GPD λ LZ1 के जीनोम और प्लाज्मिड pPlate * क्रमशः डी से सह व्यक्त की है. एक परिणाम के रूप में, नव निर्मित फेज λ LZ2 की capsid GPD EYFP और GPD प्रोटीन का एक मिश्रण होता है. इस मोज़ेक फेज structurally स्थिर और पर्याप्त फ्लोरोसेंट व्यक्ति 5 phages का पता लगाने की अनुमति है. पीपी RE - mCherry प्लाज्मिड रिपोर्टर लिए पसंद की lysogenic pathwa का पता लगाने के लिए किया जाता हैy. प्रमोटर पी पुन सीआईआई द्वारा 1,11 lysogeny की स्थापना के दौरान सक्रिय है. पीपी RE - mCherry 5 पीई GFP 11 से 12 mCherry के साथ GFP की जगह निकाली थी. अधिक जानकारी के लिए हमारे पहले 5 काम देखते हैं.
विकास की स्थिति पैरामीटर:
(खंड 1) lysogen प्रेरण के दौरान, 180 rpm पर हल्के झटकों एक अच्छा वायरस उपज 13 देता है. मध्यम विकास में ग्लूकोज का प्रयोग ग्लूकोज चयापचय के रूप में बचा जाना चाहिए अम्लीय चयापचय उत्पादों उत्पन्न करता है, और परिपक्व लैम्ब्डा कणों अम्लीय पीएच 13 में अस्थिर कर रहे हैं. 4 MgSO के अलावा फेज 3 capsid स्थिर करने के उद्देश्य से है. Phages को जंगली प्रकार CI (857 CI के बजाय) ले जाने के लिए, lysogen डीएनए हानिकारक एजेंट mitomycin सी 3 का उपयोग कर प्रेरित किया जा सकता है. 1.3 चरण में, 37 पर ऊष्मायन डिग्री सेल्सियस सामान्य रूप से 90 मिनट से अधिक नहीं होना चाहिए. यह usef है 600 आयुध डिपो से हर 30 मिनट सेल घनत्व की जांच उल. Lysate के लिए एक अच्छा, आयुध डिपो 600 0.2 के आसपास या कम करने के लिए चला जाता है, और शेष 600 आयुध डिपो सेल मलबे का एक परिणाम है. बहुत लंबे समय Incubating एक कम फेज उपज में परिणाम के बाद नव निर्मित फेज सेल मलबे में उनके डीएनए इंजेक्षन शुरू हो सकता है. 2.11 और 2.13 कदम में एक दृश्य फेज बैंड (कम से कम 1 x 10 11 फेज कण) प्राप्त करने के लिए, 1.2 चरण में कम से कम 500 मिलीलीटर संस्कृति विकसित. कदम 5.1 और 5.2 में मध्यम विकास में 0.2% Maltose के अलावा भेड़, फेज lambda 3,14 सोखना के लिए रिसेप्टर की अभिव्यक्ति उत्प्रेरण के उद्देश्य से है. MCherry 5.2 चरण में 100 गुना के बजाय 1000 गुना कमजोर पड़ने संवाददाता प्लाज्मिड पीपी RE से mCherry पृष्ठभूमि के स्तर को कम करने के उद्देश्य से है. चरण फेज डीएनए ट्रिगर, 35 इंजेक्शन के लिए 5.5 डिग्री सेल्सियस तापमान के प्रति संवेदनशील cI857 एलील की प्रेरण से बचने के लिए चुना है.
फेज शुद्धीकरण:
jove_content "> फेज शुद्धि कदम (2.11 के माध्यम से 2.1 कदम) अन्य में शुद्धि 5 प्रोटोकॉल के साथ प्रतिस्थापित किया जा सकता है, लेकिन CsCl संतुलन ढाल (कदम 2.12 और 2.13) के माध्यम से अंतिम ultracentrifugation अपरिहार्य है बाल्टी झूलते रोटार 2.10 कदम और 2.12 के लिए आवश्यक हैं. तेज दिखाई फेज बैंड सुनिश्चित एक शुद्ध फेज स्टॉक प्राप्त करने के आसानी से एक सप्ताह तक लग सकते हैं, इसलिए यह जरूरी है कि जिस तरह से साथ फेज अनुमापांक जांच करने के लिए यकीन है कि कुछ भी नहीं मध्यवर्ती कदम के दौरान गलत हो जाता है.फेज हैंडलिंग:
धारा 2 में सभी शोधन प्रक्रिया के दौरान, यह संभाल फेज धीरे lysate फेज सिर से पूंछ फेज कर्तन से बचने के लिए महत्वपूर्ण है. धारा 5 (उदाहरण के लिए, 5.5 5.7 के माध्यम से कदम) में सेल के संक्रमण के दौरान, यह भी संक्रमित कोशिका से फेज कणों की कर्तन से बचने के लिए महत्वपूर्ण है. ध्यान दें कि यदि फेज इसकी डीएनए इंजेक्शन लगाने के बाद संक्रमित कोशिका से sheared है, परिणाम एक "अंधेरे" संक्रमण है, अर्थात् मेंfection परिणाम प्रयोग में मनाया जाएगा, लेकिन को संक्रमित करने फेज नहीं होगा. इस तरह की समस्याओं को कम करने के लिए, हम एक विस्तृत विंदुक टिप का उपयोग जब भी phages या मिश्रण / फेज सेल से निपटने.
DAPI परीक्षण:
DAPI (धारा 4) के साथ फेज स्टॉक धुंधला फेज स्टॉक की शुद्धता की जांच के लिए एक त्वरित और कारगर तरीका है. यह भी समय पर एक मौजूदा फेज स्टॉक के संभावित गिरावट के लिए परीक्षण करने के लिए इस्तेमाल किया जा सकता है. स्टॉक के लिए एक शुद्ध, प्रतिदीप्ति खुर्दबीन के नीचे YFP और DAPI संकेतों के सह स्थानीयकरण 100% के करीब होना चाहिए. हम आम तौर पर पालन कि YFP स्पॉट के कम से कम 1% (वायरल जीनोम बिना capsids का प्रतिनिधित्व) DAPI है, जो बताता है कि इन कणों को सफलतापूर्वक वायरल डीएनए पैकेज नहीं किया था या पहले से ही उनके डीएनए कहीं इंजेक्शन शामिल नहीं है. DAPI स्पॉट की 1% से कम YFP (गैर फ्लोरोसेंट phages के लिए इसी) शामिल नहीं है. यदि यह मामला नहीं है, आवश्यकता के माध्यम से 2.14 2.12 कदम ओ में भी दोहराया जाको शुद्ध करने के लिए फिर से rder. इमेजिंग पैरामीटर के साथ संबंध है, 4.3 चरण में माइक्रोस्कोप सेटअप के रूप में धारा 5 में के रूप में महत्वपूर्ण नहीं है क्योंकि कोई लंबे समय तक जीवित कोशिका इमेजिंग यहाँ की आवश्यकता है. हालांकि, धारा 5 में के रूप में एक ही माइक्रोस्कोपी सेटिंग्स रखने अगर एक इच्छाओं एक सिंगल फेज कण के प्रतिदीप्ति तीव्रता जांच करने के लिए उपयोगी है. यदि पीबीएस - agarose स्लैब बहुत साफ नहीं है, या बहुत ज्यादा DAPI डाई का इस्तेमाल किया गया है, कुछ DAPI फेज डीएनए को इसी स्पॉट एक "प्रभामंडल" के साथ घिरा हुआ जा सकता है. यदि बहुत कम DAPI डाई का इस्तेमाल किया जाता है, DAPI चैनल से बहुत कमजोर संकेत हो सकता है.
माइक्रोस्कोप प्रणाली:
धारा 6 में इमेजिंग के लिए, हम एक 100x (योजना Fluo, संख्यात्मक एपर्चर 1.40, तेल विसर्जन) उद्देश्य और मानक फिल्टर सेट (Nikon) के साथ एक वाणिज्यिक औंधा epifluorescence खुर्दबीन (ग्रहण TE2000 - ई, Nikon) का उपयोग करें. प्रतिदीप्ति प्रकाश स्रोत प्रकाश की तीव्रता का नियंत्रण के साथ एक आर्क दीपक है. एक्स, वाई और z पुलिस: निम्नलिखित विशेषताएं कंप्यूटर नियंत्रित कर रहे हैंउज्ज्वल क्षेत्र और प्रतिदीप्ति बंद; sition और प्रतिदीप्ति फ़िल्टर विकल्प. एक ऑटो फोकस सुविधा की आवश्यकता है. अन्यथा, ध्यान फिल्म समय चूक (सामान्य रूप से चार घंटे लंबी) के दौरान आसानी से दूर बहाव हो सकता है. प्रत्येक समय बिंदु पर एकाधिक (एक्स, वाई) के पदों को हासिल करने की क्षमता उपयोगी है, के रूप में यह समांतर में एकाधिक संक्रमण घटनाओं का पालन करने की अनुमति देता है. आम तौर पर हम हर फिल्म में 8 चरण पदों 100 संक्रमण की घटनाओं के लिए निम्नलिखित हासिल. कैमरा का उपयोग हम 16 बिट (Cascade512, Photometrics) के एक गतिशील रेंज के साथ 16x16 सुक्ष्ममापी पिक्सेल कैमरा के साथ एक ठंडा 512x512 सीसीडी है. अधिग्रहण Metamorph सॉफ्टवेयर (आण्विक उपकरण) का उपयोग किया जाता है. एक कमरे के तापमान नियंत्रित खुर्दबीन में रखा जाना चाहिए, वैकल्पिक रूप से, मंच खुर्दबीन तापमान नियंत्रित एक कक्ष से घिरा होना चाहिए.
छवि अधिग्रहण:
जीना सेल इमेजिंग के लिए, यह महत्वपूर्ण है नमूना के अनावश्यक जोखिम से बचने के लिए, जो सफेद और फोटो के लिए नेतृत्व कर सकते हैंtotoxicity. इसलिए, यह सबसे अच्छा है पहले अपने सिस्टम विशेषताएँ एक इष्टतम प्रकाश जोखिम है जो प्रतिदीप्ति का पता लगाने के लिए अनुमति देता है विरंजन अत्यधिक या बाधा सेल के विकास के लिए नहीं, जबकि अग्रणी. एक अच्छा प्रतिदीप्ति छवि प्राप्त करने के लिए, रोमांचक प्रकाश की तीव्रता, समय जोखिम और कैमरा लाभ के साथ खेलते हैं. 6.2-6.3 चरणों में 10 मिनट फ्रेम अंतराल प्रकाश जोखिम को कम करने के उद्देश्य के लिए चुना जाता है. हर फ्रेम में, केवल एक ही छवि में फोकस चरण विपरीत (सेल की मान्यता के लिए) और फ्लोरोसेंट चैनलों (सेल भाग्य का निर्धारण करने के लिए) की जरूरत है. पहली बार बिंदु में, तथापि, YFP चैनल के माध्यम से कई z-स्थिति छवियों कोशिका की सतह पर सभी को संक्रमित करने phages पर कब्जा करने के लिए आवश्यक हैं. प्रारंभिक फ्रेम में YFP जोखिम समय भी समय व्यतीत हो जाने के बाद समय फ्रेम में फिल्म के लिए इस्तेमाल किया है कि तुलना में अधिक होने की आवश्यकता हो सकती है.
छवि विश्लेषण:
बहुत सावधानी से 7.1 चरण में सेल की सतह के आसपास फेज कणों गिनती. जैसाऊपर उल्लेख किया है, हम 6.2 चरण में YFP चैनल के माध्यम से z के ढेर की एक श्रृंखला ले. हालांकि, यह अभी भी कुछ फ्लोरोसेंट फेज बाहर का ध्यान केंद्रित कण, जो गिनती चुनौतियों को छोड़ सकते हैं. प्रारंभिक समय सीमा में सेल लंबाई Metamorph सॉफ्टवेयर का उपयोग कर मापा जाता है. सेल लंबाई भी ImageJ या अन्य सॉफ्टवेयर औजार द्वारा मापा जा सकता है. इसके अतिरिक्त, एक स्वचालित घर बनाया Matlab कार्यक्रम सेल प्रजातियों साथ समय पर प्रतिदीप्ति परिवर्तन के रूप में इस तरह की जानकारी प्राप्त करने में बहुत उपयोगी हो सकता है.
Disclosures
ब्याज की कोई संघर्ष की घोषणा की.
Acknowledgments
हम माइकल Feiss और फेज निर्माण और शोधन पर मार्गदर्शन के लिए जीन सिप्पी के लिए आभारी हैं. हम सेल मान्यता सॉफ्टवेयर, Schnitzcell प्रदान करने के लिए माइकल Elowitz धन्यवाद. वेल्श फाउंडेशन (अनुदान क्यू 1759) और मानव फ्रंटियर विज्ञान गोल्डिंग प्रयोगशाला में काम राष्ट्रीय स्वास्थ्य संस्थान (R01GM082837), राष्ट्रीय विज्ञान फाउंडेशन (जीवित कोशिकाओं के भौतिकी के लिए केंद्र 082265, पीएफसी) से अनुदान द्वारा समर्थित है प्रोग्राम (rgy 70/2008).
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma-Aldrich | D4527-10KU | |
| RNase | Sigma-Aldrich | R4642-10MG | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| SM buffer | TEKnova, Inc. | S0249 | |
| NZYM | TEKnova, Inc. | N2062 | |
| CsCl | Sigma-Aldrich | C3011-250G | |
| Syringe | BD Biosciences | 309585 | |
| Needle | BD Biosciences | 305176 | |
| Dialysis cassette | Thermo Fisher Scientific, Inc. | 66333 | |
| Microscope slide | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti ultra-clear tube | Beckman Coulter Inc. | 344060 | |
| SW60Ti ultra-clear tube | Beckman Coulter Inc. | 344062 | |
| SW40Ti rotor | Beckman Coulter Inc. | 331302 | |
| SW60Ti rotor | Beckman Coulter Inc. | 335649 | |
| Refractometer | Fisher Scientific | 13-947 | |
| Epifluorescence microscope | Nikon Instruments | Eclipse TE2000-E | |
| Table 2. Reagents and equipment. | |||
References
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S.
- Ptashne, M. A genetic switch : phage lambda revisited. , 3rd edn, Cold Spring Harbor Laboratory Press. (2004).
- Hendrix, R. W. Lambda II. , Cold Spring Harbor Laboratory. (1983).
- Hershey, A. D. The Bacteriophage lambda. , Cold Spring Harbor Laboratory. (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. Current protocols in molecular biology. , John Wiley & Sons. (1994).
- Sambrook, J., Russell, D. W. Molecular cloning : a laboratory manual. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Fasman, G. D. Practical handbook of biochemistry and molecular biology. , CRC press. (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Personal communication with M. Feiss. , Forthcoming.
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).

{kind=link}