

Summary
Este artículo describe el procedimiento para la preparación de una versión marcado fluorescentemente de bacteriófago lambda, la infección de
Abstract
El sistema comprende bacteriófago (fago) lambda y la bacteria E. coli ha servido durante mucho tiempo como un paradigma para el 1,2 determinación de la celda de destino. Después de la infección simultánea de la célula por un número de fagos, una de las dos vías se elige: lítico (virulento) o lisogénico (inactivo) 3,4. Recientemente hemos desarrollado un método para fluorescentemente etiquetado fagos individuales, y fueron capaces de examinar la decisión después de la infección en tiempo real bajo el microscopio, en el nivel de fagos individuales y las células 5. A continuación, se describe el procedimiento completo para realizar los experimentos de infección descritos en nuestro anterior trabajo 5. Esto incluye la creación de fagos fluorescentes, infección de las células, formación de imágenes en el microscopio y el análisis de datos. El fago fluorescente es un "híbrido", co-expresan versiones de tipo salvaje y YFP fusión de la proteína de la cápside GPD. Un lisado de fago en bruto se obtuvo por primera vez mediante la inducción de un lisógeno de la (GPD-EYFP Enhbrado Yellow Fluorescent Protein) de fagos, que alberga un plásmido que expresa salvaje grupo D tipo. Una serie de pasos de purificación se realiza a continuación, seguido por DAPI-etiquetado y la formación de imágenes en el microscopio. Esto se hace con el fin de verificar la uniformidad, la eficacia de empaquetamiento del ADN, la señal de fluorescencia y la estabilidad estructural del stock de fago. La adsorción inicial de fagos a las bacterias se realizaron en hielo, a continuación, seguido por una breve incubación a 35 ° C para provocar la inyección de ADN viral 6. El fago / mezcla de bacterias se mueve entonces a la superficie de una losa delgada de agar nutriente, cubierto con un cubreobjetos y fotografiado bajo un microscopio de epifluorescencia. El proceso de post-infección se siguió durante 4 horas, a intervalos de 10 min. Múltiples posiciones de fase se realiza un seguimiento de tal manera que ~ 100 infecciones de células se puede rastrear en un solo experimento. En cada punto de posición y el tiempo, las imágenes se adquieren en el contraste de fase y canales fluorescentes rojas y verdes. La imagen de contraste de fase se utiliza posteriormente para la automatización cell reconocimiento, mientras que los canales fluorescentes se utilizan para caracterizar la infección por resultado: la producción de nuevos fagos fluorescente (verde) seguido por la lisis celular, o la expresión de los factores de lisogenia (rojo) seguido por el crecimiento celular y la división se reanuda. Los adquiridos time-lapse películas se procesan utilizando una combinación de métodos manuales y automatizados. Resultados Los datos de análisis en la identificación de los parámetros de infección para cada evento infección (por ejemplo, número y posiciones de infectar a los fagos), así como resultado la infección (lisis / lisogenia). Los parámetros adicionales se puede extraer si se desea.
Protocol
1. Creación de un lisado de fago cruda (Figura 1)
- En un matraz de 50 ml, se inocula una colonia fresca de LE392 (λ LZ1) [pPLate * D] (véase la Tabla 1 para detalles) en 6 ml de medio LB suplementado con 10 7 ug / ml de kanamicina y 100 mg / ml de ampicilina. Crecer durante la noche a 30 ° C con agitación suave (180 rpm).
- Diluir la cultura 1:100 en LBM (LB suplementado con 10 mM MgSO 4) y crecen a 30 ° C con agitación suave (180 rpm). Con el fin de optimizar el rendimiento del fago, asegurarse de que el volumen de cultivo no es más que una décima parte de la capacidad de volumen del matraz. Nos típicamente preparar dos matraces de 2 litros de capacidad o 2,5 litros, y añadir 2,5 ml de cultivo durante la noche en medio LBM 250 ml en cada matraz.
- Cuando la densidad celular alcanza OD 600 ≈ 0,6 (~ 2,5 a 3 h), se provoca el lisógeno moviendo el cultivo a 42 ° C un baño de agua agitador durante 18 min con agitación suave (180 rpm) y, a continuación incue a 37 ° C con agitación suave (180 rpm) hasta que la lisis es visible (cultivo se vuelve clara, en ~ 60 - 90 min).
- Añadir 2% de cloroformo a la cultura, agitar para mezclar a mano, y luego se incuba durante 15 min a temperatura ambiente. Precaución: Use guantes para manejar el cloroformo, y evite respirar el mismo.
- Transferencia de la cultura en dos botellas de 250 ml de centrífuga, centrifugar el cultivo en un rotor Sorvall GSA a 10.000 rpm durante 15 min a 4 ° C. Recuperar el sobrenadante que contiene las partículas de fago, y desechar el residuo de restos. Realizar una segunda centrifugación para asegurarse de deshacerse de los restos visibles.
- Utilizar un protocolo de fagos de titulación estándar 8 para medir la concentración de fagos. El título de fago debe ser ~ 5-10 x 10 9 ufp / ml. Usar una cepa supF tal como LE392 como la cepa indicadora debido a la mutación SAM7 en el genotipo del fago fluorescente, y el uso de agar superior y placas de agar hechas con NZYM rica para obtener grandes placas (Figura 2).
2. Fago purificación (Figura 1)
- Verter el lisado en una grande (por ejemplo 2-litros) matraz, añadir DNasa I y RNasa (1 mg / ml cada uno) para el lisado con el fin de digerir los ácidos nucleicos liberados de las bacterias lisadas, y se incuba 1 hora a temperatura ambiente.
- Añadir NaCl 1 M al lisado, transferir el lisado en botellas de 250 ml de centrífuga, y se incuba 3 horas en hielo. Se centrifuga el lisado en un Sorvall GSA a 10.000 rpm durante 15 min a 4 ° C. Recuperar el sobrenadante. El título de fago debe ser similar a la del lisado crudo, que es ~ 5-10 x 10 9 ufp / ml. La adición de NaCl promueve la disociación de partículas de fago a partir de restos de bacterias y se requiere para la precipitación eficaz de partículas de fago por PEG 8.
- Verter el lisado en un matraz grande, por ejemplo 2-litros matraz, añadir 10% (w / v) de PEG8000 en el lisado, lentamente revolver o agitar para disolver PEG8000 a temperatura ambiente. Transferir el lisado en 250 ml cientorifuge botellas y luego incubar durante la noche (~ 16 h) a 4 ° C. Se centrifuga el lisado en un rotor Sorvall GSA a 10.000 rpm durante 15 min a 4 ° C. Eliminar el sobrenadante.
- Remojar el sedimento (partículas de fago se precipitó con PEG8000) con el fago tampón SM (4 ml de tampón SM por 250 ml de lisado de fago inicial). Incubar con agitación muy suave o sin agitación durante 16 horas a 4 ° C.
- Suavemente llevar a cabo el lisado (tampón SM con las partículas de fago) en un tubo de 50 ml de centrífuga Eppendorf, y luego lavar el sedimento remanente con 0,5 - 1 ml de tampón SM.
- Añadir un volumen igual de cloroformo al lisado. Mezclar suavemente el lisado con cloroformo hasta inversora y hacia abajo por un par de veces. Centrifugar a 4.000 rpm durante 15 min a 4 ° C en un Eppendorf 5804R o una centrífuga de sobremesa similares.
- Repita el paso 2.6 para obtener un lisado claro. El título de fago debe ser ~ 1-2 x 10 11 pfu / ml.
- Preparar SM / CsCl soluciones con tres densidades diferentes (ρ) de 1,3 g / ml, 1.5 G / ml y 1,7 g / ml. Medir el índice de refracción (η) para obtener una lectura de densidad más precisa. La conversión de densidad ρ = 9 es η 10,8601 - 13,4974 a 25 ° C. Ver Tabla 3 para obtener más información.
- Usar una jeringa con una aguja larga para cargar la solución en un 14 ml ultra-claro tubo de ultracentrífuga Beckman 40Ti. Para evitar la mezcla y para formar un gradiente de densidad mejor, como fondo para la solución (es decir, soluciones de capas de densidad creciente debajo de la otra) se debe utilizar, es decir, suavemente cargar 2 ml de SM / CsCl soluciones en el orden de 1,3 g / ml, 1,5 g / ml y 1,7 g / ml mediante la inserción de la aguja con una jeringa de 3 ml a la parte inferior del tubo.
- Suavemente cargar 8 ml de lisado de fago mediante la superposición de la parte superior del tubo de ultracentrífuga de 14 ml. Preparar un tubo de equilibrio. Se centrifuga en un rotor Beckman SW40Ti a 24.000 rpm durante 4 horas a 4 ° C.
- Con suavidad fuera del tubo en una habitación oscura y iluminar desde la parte superior del tubo contra un fondo negro usando aflashlight. La banda del fago debe ser claramente visible en la ubicación de la interfaz entre 1,3 g / ml y 1,5 g / ml SM / CsCl capas (Figura 3A). Punción a través de la pared del tubo ligeramente por debajo de la banda con una aguja de calibre 21,5 con una jeringa de 3 ml. Suavemente recoger ~ 500 l de la suspensión de fagos. El título de fago debe ser ~ 5-10 x 10 11 pfu / ml.
- Coloque la suspensión de fagos en un 4 ml ultra-claro ultracentrífuga Beckman SW60Ti tubo rotor. Llenar el tubo con 1,5 g / ml SM / solución de CsCl. Preparar un tubo de equilibrio. Se centrifuga en un rotor Beckman SW60Ti a 35.000 rpm durante 24 horas a 4 ° C.
- Repetir el mismo procedimiento que en el Paso 2,11 para recoger el fago de la banda visible. La banda debe ser visible como se muestra en la Figura 3B.
- Cargar la suspensión de fago en un cassette de membrana de diálisis (Tabla 2) y se dializa tres veces contra un volumen de 1000-veces mayor de tampón SM a 4 º C durante una duración de 3 hr, 3 hr y Overnight (~ 16 h). El propósito de la diálisis es deshacerse de CsCl presente en la suspensión de fago. El título de fago final debe ser ~ 5-10 x 10 11 pfu / ml.
3. Preparar una placa de gel de agarosa (Figura 4)
- Limpiar 6 portaobjetos de microscopio (75 x 50 mm, 1 mm de espesor) con 70% de etanol.
- Organizar 5 diapositivas y sujetarlo con cinta como se muestra en la Figura 4.
- Mezclar 0,09 g de agarosa en medio de 6 ml en un pequeño vaso de precipitados cubierto con film transparente (lo que da 1,5% de agarosa). Calentar en una placa calefactora hasta que la solución se vuelva transparente.
- Vierta la solución de agarosa en las diapositivas garantizados.
- Coloque la última diapositiva en la parte superior, evitando burbujas de aire. Coloque el peso en la parte superior y deje que se enfríe durante ~ 30 min.
- Retire los 4 diapositivas en el lado, y envolver la losa junto con las diapositivas superior e inferior con film transparente. La losa se puede almacenar a 4 ° C durante un máximo de 3 días.
4. Prueba del stock de fago purificado
- Prepararun gel de agarosa al PBS-losa tal como se describe anteriormente (Sección 3).
- Manchar el fago purificado con DAPI. Mezclar 10 l de fago (~ 1 x 10 10 pfu / ml) con 10 l de 10 mg / ml DAPI (concentración final DAPI de 5 ug / ml), incubar durante 30 min a 4 ° C o 10 min a temperatura ambiente.
- Place 1 l de la mezcla fago / DAPI en el centro de una No.1 24 x 50 mm cubreobjetos, se superponen a una pieza pequeña (~ 10 x 10 mm) de la pre-preparado PBS-agarosa. El pequeño trozo de placa de agarosa se corta con una cuchilla de afeitar después de la corredera superior en el gel sándwich se retira. Imagen de la muestra bajo el microscopio de epifluorescencia a través de los canales de YFP y DAPI. Fagos individuales deben ser visible como limitadas por difracción fluorescentes "puntos" en ambos canales (Figura 5). Utilice el mismo microscopio y configuraciones de cámara como en el paso 6.2.
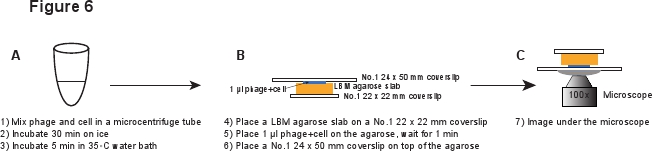
5. Infección (Figura 6)
- En un tubo de 14 ml Falcon, inocular una colonia fresca de LE392 [pp RE-MCHerry] (véase la Tabla 1 para detalles) en 2 ml de medio LB suplementado con 100 mg / ml de ampicilina, 10 mM MgSO 4 y 0,2% de maltosa. Crecer durante la noche a 37 ° C con agitación moderada (265 rpm).
- Diluir la cultura 1:1000 en LBMM (LB suplementado con 10 mM MgSO 4 y 0,2% de maltosa), es decir, añadir 5 l cultivo de una noche en 5 ml de medio LBMM en un matraz de 50 ml. Crecer a OD 600 ≈ 0,4 a 37 ° C con agitación moderada (265 rpm).
- Use el medio LBM para preparar una losa de gel de agarosa LBM-tal como se describe en la Sección 3.
- Centrifugar 1 ml de células a 2.000 g en una microcentrífuga de sobremesa durante 2 minutos a temperatura ambiente. Eliminar el sobrenadante y resuspender suavemente las células en 20 l de hielo frío LBMM para llegar a OD 600 y 20.
- Al manipular el stock de fago purificado, utilizar una pipeta de punta ancha o cortar la punta de la pipeta regular para hacer la punta de la apertura más amplia, para evitar esquilar las partículas de fago 3. Suavemente Mix 20 l de células con 20 l de fago purificado hasta alcanzar una media fagos a célula relación en el intervalo de 0,1 a 5. Incubar en hielo durante 30 min para permitir la adsorción del fago, y luego incubar en un baño de agua 35 ° C durante 5 min para activar la inyección del ADN del fago 6.
- Pipetear hacia arriba y abajo varias veces para separar las agregados de células. Una vez más utilizar una pipeta de punta ancha para evitar el cizallamiento de los fagos. Diluir la mezcla en 1:10 LBMM, por ejemplo 5 a 45 l mezcla LBMM l.
- Coloque un pedazo de losa LBM-agarosa (~ 10 x 10 mm) sobre un cubreobjetos No.1 22 mm x 22. El pre-preparado de agarosa LBM-losa debe ser colocado a temperatura ambiente durante al menos 1 hora antes de su uso para asegurarse de que la losa de agarosa alcanza la temperatura ambiente. Place 1 l de la mezcla fago / células en la losa de agarosa y esperar durante 1 minuto para permitir la mezcla para absorber en la losa de agarosa. Coloque suavemente una No.1 24 x 50 mm cubreobjetos en la parte superior de la losa de agarosa. Este procedimiento se pretende evitar cizallar los fagos de la infeja celular (Figura 6).
6. Después de destino de la célula en el microscopio
- Monte con cuidado el cubreobjetos sobre la platina del microscopio. Para las imágenes, usar un objetivo de gran aumento (por ejemplo, 100x) (véase el sistema de microscopio en adelante).
- Adquirir una imagen fijada para el período de tiempo inicial. Este conjunto de imágenes se utiliza para caracterizar los números iniciales y las posiciones de todos los fagos que infectan. Tome una serie de 15 imágenes a 200 nm eje Z (vertical) intervalos. Imagen través del canal de YFP. Además, tomar una sola imagen que tiene el foco a través del contraste de fase y canales mCherry. Optimizar la intensidad de la luz y el tiempo de exposición para obtener una señal suficiente mientras se minimiza la decoloración y el daño celular (ver Adquisición de imágenes en discusión más adelante).
- Adquirir una película de lapso de tiempo del destino celular después de la infección. Imagen de la muestra, de contraste de fase y YFP mCherrycanales a intervalos de tiempo de 10 min por alrededor de 4 horas. Durante la película de lapso de tiempo, utilizar un único z-posición de la imagen por canal, por punto de tiempo, para evitar la exposición innecesaria de la muestra, lo que podría conducir a la decoloración y la fototoxicidad.
7. Análisis de la imagen
- Manualmente contar el número de fagos y anote la localización de fagos y la longitud de la célula en el período de tiempo inicial. Esto se puede hacer usando software tal como MetaMorph o ImageJ. Registre los destinos celulares (lítico, lisogénico o no infectada), el tiempo de lisis, y cualquier otra información deseada a reproducir la película de lapso de tiempo. Para identificar los diferentes destinos celulares, vea Time-lapse películas en la sección Resultados Representante a continuación.
- Además del análisis manual anteriormente, la información más cuantitativa (por ejemplo, nivel de fluorescencia con el tiempo en las células individuales) se puede extraer utilizando automatizados de reconocimiento de células linaje y algoritmos de búsqueda. Utilizamos una casa construida programa Matlab para tracing el linaje de células y los niveles de fluorescencia, junto con el código Schnitzcell Matlab para la segmentación celular (escrito por el grupo Elowitz en Caltech).
8. Los resultados representativos:
Fago Plating:
Las placas de los fagos marcados con fluorescencia (en 1,6 y en la sección Paso 2) son significativamente más pequeños que los de tipo salvaje (Figura 2). Por lo tanto, se incuban las placas de al menos 12 horas en 37 ° C incubadora para las placas a ser visible.
Fago Ultracentrifugación:
Después de la ultracentrifugación de la muestra de fagos con el gradiente escalonado de CsCl (paso 2,10), dos bandas deben ser visibles (Figura 3A). La banda superior, en la interfaz entre la suspensión de fago y SM / CsCl 1,3 capa g / ml, contiene restos de células y cápsidas vacías de fagos. La banda inferior, en la interfaz entre SM / CsCl 1,3 g / ml y 1,5 capas g / ml, es la banda de fagos. This banda aparece verdosa fluorescente para el fago λ LZ2. La banda de tipo salvaje fago λ IG2903 aparece azulado 5. Después de la ultracentrifugación en gradiente de equilibrio de CsCl en el paso 2,12, una banda de fago debe ser visible en la parte media del tubo (Figura 3B). Dado que el fago λ LZ2 fluorescente contiene una mezcla de GPD-EYFP y GPD cápsidas, la relación de proteína a ADN es mayor que la de tipo salvaje. Por lo tanto, la banda de la fluorescente fago λ LZ2 es ligeramente más claro (parece estar en un lugar más alto en el tubo) que el de tipo salvaje λ IG290310.
Tinción DAPI:
La Figura 5 muestra imágenes típicas obtenidas después de marcar el fago con DAPI (Sección 4). Los YFP y DAPI señales de un fago purificado con éxito debe tener cerca de 100% correspondencia. Nos típicamente observar que menos del 1% de la mancha YFPs no contienen DAPI (representando cápsidas sin el genoma viral). Menos del 1% de los puntos DAPI no contienen YFP (correspondiente a los fagos no fluorescentes) 5.
Time-lapse película:
Células líticas son reconocidos por la acumulación de YFP fluorescencia (canal verde) dentro de la célula, seguido por la lisis celular. Células lisogénicas son reconocidos por la acumulación de fluorescencia uniforme mCherry (rojo) en el interior de la célula y la reanudación del crecimiento celular normal y división. Las células no infectadas (o células donde la infección no ha funcionado) no presenta ninguna de los fenotipos anteriores y crecen y se dividen normalmente. Figura 7 muestra unas pocas imágenes-conjuntos de contraste de fase, YFP y canales mCherry, y las imágenes correspondientes superpuestas de estos tres canales, de una típica película de lapso de tiempo (Sección 6). Los fagos individuales (manchas verdes) son claramente visibles en el marco de tiempo inicial (Figura 7A). Normalmente, un númerofagos de se ven en la superficie celular (presumiblemente infectar dichas células), mientras que otros fagos son no adsorbido, como se muestra en la Figura 7B (panel izquierdo). El resultado se convierte en infección distinguible en el tiempo. El ciclo lítico se indica mediante la producción intracelular de nuevos fagos (verde, Figura 7C), seguido por la lisis celular (células explotaron con fagos liberados verdes, Figura 7D). Lisogenia se indica mediante la producción de mCherry a partir del promotor P RE (rojo, Figura 7C) y la reanudación del crecimiento y división celular (rojo, Figura 7D).




Figura 1. AB). El fago se purificó a través de una serie de pasos (CL paneles).

Figura 2. Placas de fago. Las placas de los fagos fluorescente (a la izquierda) son más pequeños que los de tipo salvaje (a la derecha) después de incubar las placas durante 12 horas a 37 ° C.

Figura 3. Bandas de fagos después de la ultracentrifugación. A) Dos bandas son visibles después de la ultracentrifugación en un gradiente escalonado de CsCl. La de arriba corresponde a los restos celulares y cápsides vacías de fagos, y la banda inferior contiene el fago deseado. Izquierda: fago fluorescente, derecha:. Tipo silvestre B ) Una banda de fago solo es visible después de la ultracentrifugación en un gradiente de equilibrio de CsCl. La banda fago fluorescente (a la izquierda) es de color verdoso, en comparación con una banda azul de fago de tipo salvaje (derecha).

Figura 4. El procedimiento de preparación de placas de gel de agarosa.

Figura 5. Imágenes fluorescentes de fagos después de la tinción DAPI. Fagos individuales son fácilmente distinguibles, y YFP y DAPI señales co-localizar muy bien.

Figura 6. Descripción esquemática de la infección por fagos y la configuración de imagen. Haga clic aquí para ver una versión en tamaño completo de la imagen.
gura 7 "src =" / files/ftp_upload/3363/3363fig7.jpg "/>
Figura 7. Imágenes típicas de una película de lapso de tiempo de la infección por fagos. Se muestra el contraste de fase, YFP y canales mCherry, así como una superposición de los tres canales. (A) YFP canales imágenes de la período de tiempo inicial. Izquierda, la suma de las imágenes en diferentes YFP z posiciones. Las tres imágenes de la derecha son imágenes de muestra YFP en diferentes z posiciones, correspondientes a las diferentes áreas de la superficie de la célula. (B), (C) y (D) las imágenes superpuestas (izquierda) de la fase de contraste (centro-izquierda), YFP (centro-derecha) y mCherry (derecha) canales en distintos marcos temporales. (B) En t = 0, dos células se ven, cada infectada por un fago solo (manchas verdes), y una célula es infectada por 3 fagos. También se observan algunos fagos adsorbidos. (C) En t = 80 min, las dos células infectadas por fagos individuales han seguido cada una en la vía lítica, como indicand por la producción intracelular de nuevos fagos (verde). La célula infectada por 3 fagos ha entrado en la vía lisogénica, como se indica por la producción de mCherry a partir del promotor PRE (rojo). (D) En t = 2 h, la vía lítica ha dado lugar a la lisis celular (células de despiece), mientras que la célula lisogénica ha dividido §.
§ Paneles izquierdo de la Figura 7 (C) y (D) son extraídas de la célula, 141, Lanying Zeng, Samuel O. Skinner, Zong Chenghang, Sippy Jean, Feiss Michael, y Golding Ido, toma de decisiones a nivel subcelular determina el resultado de la infección por bacteriófagos, 682-691, Copyright (2010), con permiso de Elsevier.
| Colar nombre | Genotipo relevantes | Fuente / referencia |
| Cepas bacterianas | ||
| LE392 | cenarF | John Cronan, Universidad de Illinois |
| Cepas de fagos | ||
| λ LZ1 | GPD-EYFP, cI857 SAM7 D-EYFP b :: kanR | Zeng et al. 5 |
| λ LZ2 | GPD-mosaico, mismo genotipo que λ LZ1 | Zeng et al. 5 |
| Los plásmidos | ||
| pP RE - mCherry | mCherry bajo el control de P RE, R amp | Zeng et al. 5 |
| pPLate * D | grupo D bajo el control del promotor λ tarde, amp R | Zeng et al. 5 |
Tabla 1. Cepas bacterianas,fagos y plásmidos utilizados en este trabajo.
| Densidad ρ (g / ml) | CsCl (g) | SM (ml) | Indice de refracción η |
| 1,30 | 39 | 86 | 1,3625 |
| 1,50 | 67 | 82 | 1,3815 |
| 1,70 | 95 | 75 | 1,3990 |
Tabla 3. Soluciones de CsCl preparada en tampón SM (100 ml) para gradientes escalonados.
Discussion
Cepas bacterianas, fagos y plásmidos:
La cepa LE392 es supF. Fue elegida para suprimir la mutación SAM7 en el genoma del fago (véase la Tabla 1 para detalles). Por lo tanto, lisógenos inducidos finalmente lisis y libera partículas de fagos, como células infectadas que han optado por la vía lítica. Células lisogénicas se cultivan a 30 ° C debido a la presencia de la temperatura cI sensible alelo 857 en el genoma del fago. Después de la inducción de calor, GPD-EYFP y de tipo salvaje grupo D se co-expresan a partir del genoma de λ LZ1 y la pPlate plásmido * D respectivamente. Como resultado, la cápside de la recién creada LZ2 fago λ contiene una mezcla de GPD-EYFP y proteínas GPD. Este fago mosaico es estructuralmente estable y fluorescente suficientemente para permitir la detección de fagos individuales 5. pP RE - mCherry es un plásmido utilizado para detectar elección de la pathwa lisogénicoy. El RE promotor P se activa por CII durante el establecimiento de la lisogenia 1,11. pP RE - mCherry 5 se deriva de pE-gfp 11 mediante la sustitución de las buenas prácticas agrarias con mCherry 12. Para más detalles vea nuestro trabajo anterior 5.
Parámetros de Crecimiento Estado:
Durante la inducción lisógeno (Sección 1), agitación suave a 180 rpm da un rendimiento bueno virus 13. El uso de la glucosa en el medio de crecimiento debe ser evitado como metabolismo de la glucosa genera productos metabólicos ácidos, y partículas maduras lambda son inestables a pH ácido 13. La adición de MgSO 4 está dirigido a la estabilización de la cápside del fago 3. Para los fagos portadores de tipo salvaje cI (en lugar de cI 857), el lisógeno puede ser inducida mediante el C daña el ADN Mitomicina agente 3. En el paso 1.3, la incubación a 37 ° C no debe ser superior a 90 minutos. Es USEF ul para comprobar la densidad celular por OD 600 cada 30 min. Por un lisado buena, OD 600 cae a alrededor de 0,2 o menos, y el restante OD 600 es un resultado de los restos celulares. Incubando durante demasiado tiempo puede resultar en un rendimiento inferior fago desde el fago recién creada puede comenzar a inyectar su ADN en los restos celulares. Para obtener una banda de fago visible (por lo menos 1 x 10 11 partículas de fago) en los pasos 2,11 y 2,13, crecer al menos 500 ml de cultivo en el paso 1,2. La adición de 0,2% de maltosa en el medio de crecimiento en los Pasos 5,1 y 5,2 está dirigido a inducir la expresión de cordero, el receptor para el fago lambda adsorción 3,14. La dilución de 1000 veces en lugar de 100 veces en el paso 5.2 está dirigida a reducir el nivel de fondo de la mCherry RE reportero plásmido pP - mCherry. En el paso 5,5 para la inyección de ADN de fago de activación, 35 ° C se elige para evitar la inducción del alelo cI857 sensible a la temperatura.
Fago Purificación:
jove_content "> Las etapas de purificación de fagos (Pasos 2,1 a través de 2,11) se puede reemplazar con otros protocolos de purificación 5, pero el final a través de ultracentrifugación en gradiente de equilibrio de CsCl (Pasos 2,12 y 2,13) es inevitable. Rotores basculantes son necesarios en los pasos 2,10 y 2,12 a asegurar afilados bandas de fagos visibles. Obtención de un stock de fago puro puede tardar hasta una semana, por lo que es necesario comprobar el título del fago en el camino para asegurarse de que nada va mal durante los pasos intermedios.Fago Manipulación:
Durante todos los procedimientos de purificación en la Sección 2, que es crítico para manejar fago lisado suavemente para evitar la cizalladura colas de fago a partir de cabezas de fagos. Durante la infección de células en la Sección 5 (por ejemplo, a través de los pasos 5,5 5,7), también es crítica para evitar el cizallamiento de las partículas de fagos de la célula infectada. Tenga en cuenta que si el fago se cizalla de la célula infectada después de inyectar su ADN, el resultado es un "oscuro" infección, es decir, en lafection resultado se observó en el experimento pero el fago infecta no. Para minimizar tales problemas, se usa una punta de pipeta de ancho cuando el manejo de los fagos o de la mezcla fago / células.
DAPI Pruebas:
La tinción de los stock de fago con DAPI (Sección 4) es un método rápido y eficiente para comprobar la pureza de la población de fagos. También se puede utilizar para comprobar la posible degradación de un stock de fago existente en el tiempo. Para una acción pura, la co-localización de las señales de YFP y DAPI bajo el microscopio de fluorescencia debe ser cercana a 100%. Nos típicamente observar que menos del 1% de los puntos YFP no contienen DAPI (representando cápsidas sin el genoma viral), lo que indica que estas partículas no con éxito empaquetar el ADN viral o había inyectado ya su ADN en otro lugar. Menos del 1% de los puntos DAPI no contienen YFP (correspondiente a los fagos no fluorescentes). Si este no es el caso, a través de los pasos 2,12 2,14 necesidad a repetirse en OrDer para purificar de nuevo. Con respecto a los parámetros de imagen, la configuración de microscopio en el Paso 4,3 no es tan crítica como en la Sección 5 porque no a largo plazo de células vivas de imágenes se requiere aquí. Sin embargo, mantener la configuración de microscopía mismos como en la sección 5 es útil si se quiere calibrar la intensidad de fluorescencia de una partícula de fago único. Si la losa de PBS-agarosa no está muy limpio, o demasiado DAPI colorante se utiliza, algunos puntos correspondientes a DAPI ADN del fago puede estar rodeado de un "halo". Si tinte DAPI se usa demasiado poco, la señal del canal de DAPI puede ser muy débil.
Microscopio Sistema:
Para la formación de imágenes en la sección 6, se utiliza un microscopio de epifluorescencia invertida comercial (Eclipse TE2000-E, Nikon) con un objetivo de 100x (Plan Fluo, apertura numérica 1,40, aceite de inmersión) y conjuntos de filtros estándar (Nikon). La fuente de luz fluorescente es una lámpara de arco con control de intensidad de luz. Las siguientes funciones están controladas por ordenador: x, y y z poción, campo claro y fluorescencia persianas y selección de filtro de fluorescencia. Una función de enfoque automático se requiere. De lo contrario, el enfoque puede fácilmente desviarse durante la película de lapso de tiempo (normalmente de 4 horas de duración). La capacidad de adquirir múltiples posiciones (x, y) en cada punto de tiempo es útil, ya que permite a los acontecimientos múltiples de infección en paralelo. Por lo general adquieren 8 posiciones de escena en cada película, después de un máximo de 100 eventos de infección. La cámara que utilizamos es una solución enfriada CCD con 512x512 píxeles de la cámara de 16x16 micras con un rango dinámico de 16 bits (Cascade512, Fotometría). La adquisición se realizó utilizando el software Metamorph (Molecular Devices). El microscopio debe ser colocado en una habitación con temperatura controlada, como alternativa, la platina del microscopio debe ser rodeado por una cámara de temperatura controlada.
Adquisición de imágenes:
En vivo imágenes de células, es fundamental para evitar la exposición innecesaria de la muestra, lo que podría conducir a la decoloración y phototoxicity. Por lo tanto, es mejor para caracterizar primero el sistema para encontrar una exposición óptima de la luz que permite la detección de fluorescencia, mientras que no conduce a un crecimiento celular excesivo blanqueamiento o inhibición. Para obtener una imagen de fluorescencia bueno, jugar con la intensidad de la luz excitante, tiempo de exposición y ganancia de la cámara. En los pasos 6.2-6.3, el intervalo de 10 min marco se elige con el fin de minimizar la exposición de la luz. En cada trama, sólo un único foco en-imagen que se necesita en contraste de fase (para el reconocimiento de células) y los canales fluorescentes (para determinar el destino de células). En el primer punto de tiempo, sin embargo, múltiples posición z imágenes a través del canal de YFP son necesarios para capturar todos los fagos que infectan en la superficie celular. El tiempo de exposición YFP en el marco inicial también puede necesitar ser mayor que el utilizado para la película de lapso de tiempo en los marcos de tiempo posteriores.
Análisis de imagen:
Muy cuidadosamente contar partículas de fago alrededor de la superficie de la célula en el paso 7,1. Comose señaló anteriormente, tomamos una serie de z-pilas a través del canal YFP en el Paso 6.2. Sin embargo, esto puede hacer que algunas partículas de fago fluorescentes fuera de foco, que pone a prueba el recuento. La longitud de la célula en el período de tiempo inicial se mide utilizando el software Metamorph. La longitud de la célula también se puede medir por ImageJ o otras herramientas de software. Adicionalmente, un hogar automatizado construido programa Matlab puede ser muy útil en la obtención de información tales como el cambio de fluorescencia con el tiempo a lo largo de los linajes celulares.
Disclosures
No hay conflictos de interés declarado.
Acknowledgments
Damos las gracias a Michael y Jean Feiss Sippy para la orientación en la creación de fagos y purificación. Damos las gracias a Michael Elowitz para proporcionar el software de reconocimiento celular, Schnitzcell. El trabajo en el laboratorio de Golding es apoyado por becas de los Institutos Nacionales de Salud (R01GM082837), la National Science Foundation (082.265, PFC: Centro para la Física de las células vivas), la Fundación Welch (Grant Q-1759) y Ciencia Human Frontier Programa (PRG 70/2008).
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma-Aldrich | D4527-10KU | |
| RNase | Sigma-Aldrich | R4642-10MG | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| SM buffer | TEKnova, Inc. | S0249 | |
| NZYM | TEKnova, Inc. | N2062 | |
| CsCl | Sigma-Aldrich | C3011-250G | |
| Syringe | BD Biosciences | 309585 | |
| Needle | BD Biosciences | 305176 | |
| Dialysis cassette | Thermo Fisher Scientific, Inc. | 66333 | |
| Microscope slide | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti ultra-clear tube | Beckman Coulter Inc. | 344060 | |
| SW60Ti ultra-clear tube | Beckman Coulter Inc. | 344062 | |
| SW40Ti rotor | Beckman Coulter Inc. | 331302 | |
| SW60Ti rotor | Beckman Coulter Inc. | 335649 | |
| Refractometer | Fisher Scientific | 13-947 | |
| Epifluorescence microscope | Nikon Instruments | Eclipse TE2000-E | |
| Table 2. Reagents and equipment. | |||
References
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S.
- Ptashne, M. A genetic switch : phage lambda revisited. , 3rd edn, Cold Spring Harbor Laboratory Press. (2004).
- Hendrix, R. W. Lambda II. , Cold Spring Harbor Laboratory. (1983).
- Hershey, A. D. The Bacteriophage lambda. , Cold Spring Harbor Laboratory. (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. Current protocols in molecular biology. , John Wiley & Sons. (1994).
- Sambrook, J., Russell, D. W. Molecular cloning : a laboratory manual. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Fasman, G. D. Practical handbook of biochemistry and molecular biology. , CRC press. (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Personal communication with M. Feiss. , Forthcoming.
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).

{kind=link}