

Summary
Dieser Artikel beschreibt das Verfahren zur Herstellung eines fluoreszenzmarkierten Version des Bakteriophagen Lambda, Infektion
Abstract
Das System umfasst Bakteriophagen (Phage) Lambda und das Bakterium E. coli ist seit langem als ein Paradigma für die Zell-Schicksal Bestimmung 1,2 serviert. Nach der simultanen Infektion der Zelle durch eine Anzahl von Phagen wird einer der zwei Pfade ausgewählt lytischen (virulenten) oder lysogenen (ruhenden) 3,4. Wir haben kürzlich eine Methode entwickelt, Fluoreszenzmarkierung einzelnen Phagen und konnten die nach der Infektion Entscheidung in Echtzeit unter dem Mikroskop zu untersuchen, auf der Ebene der einzelnen Phagen und Zellen 5. Hier beschreiben wir die ganze Prozedur für die Durchführung der Infektion Experimente in unseren früheren Arbeiten 5 beschrieben. Dazu gehören die Schaffung von fluoreszierenden Phagen, Infektion der Zellen, Bildgebung unter dem Mikroskop und Datenanalyse. Das fluoreszierende Phage ist ein "Hybrid"-, Co-exprimierenden Wildtyp und YFP-fusion Versionen des Kapsids gpD Protein. Eine grobe Phagenlysat wird zunächst durch Induktion einer Lysogen der gpD-EYFP (Enh erhaltenWEITERFÜHRENDE Yellow Fluorescent Protein) Phagen, ein Plasmid, die Wildtyp-gpD. Eine Reihe von Reinigungsschritte werden dann durchgeführt, gefolgt von DAPI-Kennzeichnung und Bildgebung unter dem Mikroskop. Dies wird getan, um die Gleichmäßigkeit, DNA Verpackungseffizienz, Fluoreszenzsignal und strukturelle Stabilität des Phagen Lager verifizieren. Die anfängliche Adsorption der Phagen an Bakterien wird auf Eis durchgeführt, dann gefolgt von einer kurzen Inkubation bei 35 ° C, um virale DNA-Injektion 6 auslösen. Die Phagen / Bakterien Gemisch wird dann auf die Oberfläche einer dünnen Nähr-Agar-Platte, bedeckt mit einem Deckglas und abgebildet unter einem Epifluoreszenzmikroskop bewegt. Die nach der Infektion Prozess wird für 4 Stunden folgte, bei 10 min Intervall. Mehrstufige Positionen nachgeführt, so dass ~ 100 Zellen Infektionen in einem einzigen Experiment zurückgeführt werden kann. An jeder Position und Zeitpunkt werden die Bilder in der Phasenkontrast-und rot und grün fluoreszierendes Kanäle erfasst. Die Phasenkontrast-Bild wird später für die automatisierte cel verwendetl Anerkennung während die fluoreszierenden Kanäle verwendet werden, um die Infektion zu charakterisieren Ergebnis: Herstellung neuer fluoreszierenden Phagen (grün) durch Zelllyse oder Expression Lysogenie Faktoren (rot) durch Wiederaufnahme Zellwachstum und-teilung folgt. Die erworbenen Zeitraffer-Filme verarbeitet werden mit einer Kombination aus manuellen und automatisierten Methoden. Datenanalyse führt zur Identifizierung einer Infektion Parameter für jeden Infektion Ereignis (z. B. Anzahl und Positionen der infizierenden Phagen) sowie Infektion Ergebnis (Lyse / Lysogenie). Zusätzliche Parameter extrahiert werden, falls gewünscht.
Protocol
Ein. Schaffung eines rohen Phagenlysat (Abbildung 1)
- In einem 50 ml Kolben, impfen einen frischen Kolonie LE392 (λ LZ1) [pPLate * D] (siehe Tabelle 1 für weitere Details) in 6 ml LB-Medium 7 mit 10 ug / ml Kanamycin und 100 ug / ml Ampicillin ergänzt. Wachsen bei 30 ° C über Nacht mit leichten Schütteln (180 rpm).
- Verdünnen Sie die Kultur 1:100 in LBM (LB mit 10 mM MgSO 4 ergänzt) und wachsen bei 30 ° C mit mildem Schütteln (180 rpm). Um die Phagen-Ausbeute zu optimieren, um sicherzustellen, dass das Kulturvolumen nicht mehr als ein Zehntel des Kolbens Volumenkapazität ist. Wir in der Regel bereiten zwei Flaschen mit 2-Liter-oder 2,5-Liter Hubraum und 2,5 ml Übernachtkultur in 250 ml LBM in jedem Kolben.
- Wenn die Zelldichte OD600 ≈ 0.6 erreicht (~ 2,5 - 3 Stunden), induzieren die Lysogen durch Bewegen der Kultur auf eine 42 ° C-Wasserbad Schüttler für 18 min mit leichtem Schütteln (180 UpM) und dann INCUBATe bei 37 ° C mit leichter Schütteln (180 rpm), bis Lyse sichtbar ist (Kultur wird deutlich, in ~ 60 - 90 min).
- Fügen Sie 2% Chloroform zur Kultur, von Hand zu mischen schütteln, und dann inkubieren für 15 Minuten bei Raumtemperatur. Achtung: Handschuhe tragen, um Chloroform zu behandeln und zu vermeiden atmen sie.
- Übertragung der Kultur in zwei 250 ml Zentrifugenbecher, zentrifugieren die Kultur in einem Sorvall GSA-Rotor bei 10.000 UpM für 15 min bei 4 ° C. Wiederherstellen der Überstand mit den Phagenpartikel, und entsorgen Sie die Tablette aus Schutt. Führen Sie eine zweite Zentrifugation sicherstellen, um loszuwerden, der sichtbaren Verschmutzungen.
- Verwenden Sie ein Standard-Phagen Titration Protokolls Nr. 8 zum Phagen-Konzentration zu messen. Die Phagen-Titer sollte ~ 5-10 x 10 9 pfu / ml. Verwenden eines supF Stamm, wie LE392 als Indikatorstammes wegen der Sam7 Mutation in dem Genotyp des fluoreszierenden Phagen, und verwenden oberen Agar und Agar-Platten mit reicher gemacht NZYM größere Plaques zu erhalten (FiAbbildung 2).
2. Phagen Aufreinigung (Abbildung 1)
- Gießen des Lysats in eine große (zB 2-Liter) überführt und DNase I und RNase (1 pg / ml) zu dem Lysat, um die freigesetzten Nukleinsäuren aus lysierten Bakterien verdauen, und inkubieren 1 Stunde bei Raumtemperatur.
- Fügen 1M NaCl zum Lysat, übertragen das Lysat in 250 ml Zentrifuge Flaschen und inkubieren 3 h auf Eis. Zentrifuge das Lysat in einem Sorvall GSA bei 10.000 UpM für 15 min bei 4 ° C. Recover den Überstand. Die Phagen-Titer sollte ähnlich dem des rohen Lysats, welches ~ 5-10 x 10 9 pfu / ml. Die Zugabe von NaCl fördert Dissoziation von Phagenpartikeln aus bakteriellen Ablagerungen und ist für eine effiziente Fällung von Phagenpartikeln durch PEG 8 erforderlich.
- Gießen Sie das Lysat in einer großen Flasche, zB 2-Liter-Kolben, 10% (w / v) PEG8000 in dem Lysat, langsam rühren oder schütteln, um PEG8000 bei Raumtemperatur zu lösen. Übertragen Sie das Lysat in 250 ml Prozentrifuge Flaschen und dann Inkubieren über Nacht (~ 16 Stunden) bei 4 ° C. Zentrifuge das Lysat in einem Sorvall GSA-Rotor bei 10.000 UpM für 15 min bei 4 ° C. Überstand verwerfen.
- Einweichen des Pellets (Phagenpartikel ausgefällt mit PEG8000) mit Phagen SM-Puffer (4 ml SM-Puffer pro 250 ml anfänglichen Phagenlysat). Inkubieren mit sehr milden Schütteln oder kein Schütteln für 16 Stunden bei 4 ° C.
- Vorsichtig nehmen Sie das Lysat (SM-Puffer mit den Phagenpartikel) in einem 50 ml Eppendorf-Zentrifugenröhrchen, und dann waschen Sie die verbleibende Pellet mit 0,5 - 1 ml SM-Puffer.
- Hinzufügen gleichen Volumen Chloroform zum Lysat. Vorsichtig mischen das Lysat mit Chloroform durch Invertieren oben und unten für ein paar mal. Zentrifuge bei 4.000 rpm für 15 min bei 4 ° C in einem Eppendorf 5804R oder einer ähnlichen Tischzentrifuge.
- Wiederholen Sie Schritt 2,6, um ein klareres Lysat erhalten. Die Phagen-Titer sollte ~ 1-2 sein x 10 11 pfu / ml.
- Planen SM / CsCl-Lösungen mit drei unterschiedlichen Dichten (ρ) von 1,3 g / ml, 10,5 g / ml und 1,7 g / ml. Messung des Brechungsindex (η), um eine genauere Dichtewert zu erhalten. Die Dichte Umwandlung 9 ist ρ = 10,8601 η - 13,4974 bei 25 ° C. Siehe Tabelle 3 für weitere Einzelheiten.
- Verwenden Sie eine Spritze mit langer Nadel, um die Lösung in ein 14 ml ultra-clear Beckman 40Ti Ultrazentrifuge Rohr laden. Um zu vermeiden, Mischen und eine bessere-Dichtegradienten zu bilden, das Unterlegen Lösung (dh Lösungen von Schichtung zunehmender Dichte unter einem anderen) eingesetzt werden sollte, das heißt, leicht geladen werden 2 ml SM / CsCl-Lösungen in einer Größenordnung von 1,3 g / ml, 1,5 g / ml und 1,7 g / ml durch Einsetzen der Nadel mit einer 3-ml-Spritze zur Unterseite des Rohres.
- Schonend geladen werden 8 ml Phagenlysat durch Überlagerung von der Oberseite des Rohres 14 ml Ultrazentrifuge. Bereiten Sie eine Balance Rohr. Zentrifuge in einer Beckman SW40Ti Rotor bei 24.000 rpm für 4 Stunden bei 4 ° C.
- Vorsichtig nehmen Sie das Rohr in einem dunklen Raum und beleuchten aus dem oberen Ende des Rohres gegen einen schwarzen Hintergrund mit aflashlight. Die Phagen-Band sollte deutlich sichtbar am Ort der Schnittstelle zwischen 1,3 g / ml und 1,5 g / ml SM / CsCl Schichten (Abbildung 3A). Punktion durch die Seite des Rohres leicht unter dem Band mit einer 21,5-Gauge-Nadel mit einer 3-ml-Spritze. Vorsichtig sammeln ~ 500 ul der Phagensuspension. Die Phagen-Titer sollte ~ 5-10 x 10 11 pfu / ml.
- Legen Sie die Phagensuspension in ein 4 ml ultra-clear Beckman Ultrazentrifuge SW60Ti Rotorrohrs. Füllen Sie den Schlauch mit 1,5 g / ml SM / CsCl-Lösung. Bereiten Sie eine Balance Rohr. Zentrifuge in einer Beckman SW60Ti Rotor bei 35.000 rpm für 24 Stunden bei 4 ° C.
- Wiederholen des gleichen Verfahrens wie in Schritt 2,11 bis die Phagen aus dem sichtbaren Band zu sammeln. Die Bande sichtbar sein soll, wie in 3B gezeigt.
- Legen Sie die Phagensuspension in eine Dialysemembran Kassette (Tabelle 2) und Dialyse drei Mal gegen eine 1000-fache Volumen von SM-Puffer bei 4 ° C für die Dauer von 3 Stunden, 3 Stunden und overnight (~ 16 h). Der Zweck der Dialyse ist loswerden CsCl in der Phagensuspension vermischt. Die endgültige Phagentiter sollte ~ 5-10 x 10 11 pfu / ml.
3. Bereiten Sie ein Agarosegel Platte (Abb. 4)
- Sauberkeit 6 Objektträger (75 x 50 mm, 1 mm dick) mit 70% Ethanol.
- Vereinbaren 5 Rutschen und sichern mit Klebeband, wie in Abbildung 4 dargestellt.
- Mischen 0,09 g Agarose in 6 ml Medium in einem kleinen Becherglas mit Frischhaltefolie (ergibt 1,5% Agarose) abgedeckt ist. Erhitzen auf einer heißen Platte, bis die Lösung klar.
- Gießen Sie die Agarose-Lösung auf die gesicherten Dias.
- Legen Sie die letzte Folie auf der Oberseite vorsichtig Luftblasen vermeiden. Ort Gewicht auf und lassen Sie sie für ~ 30 min abkühlen.
- Entfernen Sie die 4 Rutschen auf der Seite, und wickeln Sie die Platte zusammen mit den oberen und unteren Folien mit Frischhaltefolie. Die Platte kann bei 4 ° C für bis zu 3 Tage gelagert werden.
4. Testen des gereinigten Phagen Lager
- Vorbereitena PBS-Agarosegel Platte wie oben beschrieben (Abschnitt 3).
- Flecken auf der gereinigten Phagen mit DAPI. Mix 10 ul des Phagen (~ 1 x 10 10 pfu / ml) mit 10 ul von 10 pg / ml DAPI (final DAPI Konzentration von 5 mg / ml) für 30 min bei 4 ° C inkubieren oder 10 min bei Raumtemperatur.
- Ort 1 ul des Phagen / DAPI Mischung bei der Mitte eines No.1 24 x 50 mm Deckglas, Overlay ein kleines Stück (etwa 10 x 10 mm) der vorbereiteten PBS-Agarose Bramme. Das kleine Stück Agarose Bramme mit einer Rasierklinge nach der oberen Folie auf der Sandwich-Gel entfernt geschnitten. Bild der Probe unter der Epifluoreszenzmikroskop durch die YFP und DAPI-Kanäle. Individuelle Phagen sichtbar sein soll als beugungsbegrenzte fluoreszierend "spots" in beiden Kanälen (Abbildung 5). Verwenden Sie den gleichen Mikroskop und Kamera-Setups wie in Schritt 6,2 unten.
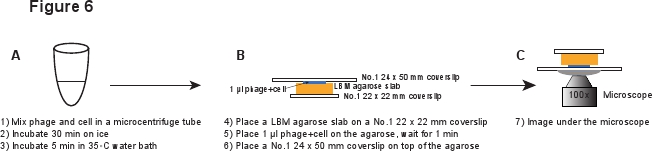
5. Infektion (Abbildung 6)
- In einem 14 ml-Falcon-Röhrchen, impfen einen frischen Kolonie LE392 [pP RE-mCherry] (siehe Tabelle 1 für weitere Details) in 2 ml LB-Medium mit 100 ug / ml Ampicillin, 10 mM MgSO 4 und 0,2% Maltose ergänzt. Wachsen bei 37 ° C über Nacht unter mäßigem Schütteln (265 rpm).
- Verdünnen Sie die Kultur 1:1000 in LBMM (LB mit 10 mM MgSO 4 und 0,2% Maltose ergänzt), fügen dh 5 ul-Nacht-Kultur in 5 ml LBMM Medium in einer 50 ml Flasche. Wachsen auf OD 600 ≈ 0,4 bei 37 ° C unter mäßigem Schütteln (265 rpm).
- Verwenden LBM Medium, um eine LBM-Agarosegel Platte vorbereiten, wie in Abschnitt 3 beschrieben.
- Zentrifuge 1 ml Zellen bei 2.000 g in einer Tischzentrifuge Mikrozentrifuge für 2 min bei Raumtemperatur. Entfernen Sie den Überstand und sanft werden die Zellen in 20 ul eiskaltem LBMM auf OD 600 und 20 zu erreichen.
- Bei der Manipulation der gereinigten Phagen Lager, nutzen ein breites Pipettenspitze oder schneiden Sie die regelmäßigen Pipettenspitze, um die Bahnöffnung breiter, um zu vermeiden Abscheren der Phagenpartikel 3. Gently Mix 20 ul Zellen mit 20 ul von gereinigtem Phagen, um eine durchschnittliche Phagen-zu-Zell-Verhältnis im Bereich von 0,1 zu erreichen - 5. Inkubieren auf Eis für 30 min auf Phagen Adsorption zu ermöglichen, und dann Inkubieren in einem 35 ° C warmen Wasserbad 5 Minuten lang, um Phagen-DNA Einspritzung 6 auslösen.
- Und Abpipettieren ein paar Mal keine Zellaggregate trennen. Wieder verwenden eine breite Pipettenspitze zu vermeiden Abscheren der Phagen. Verdünnen Sie die Mischung 1.10 in LBMM, zB 5 ul Mischung in 45 ul LBMM.
- Legen Sie ein Stück LBM-Agarose Platte (~ 10 x 10 mm) auf einem No.1 22 x 22 mm Deckglas. Die vorbereiteten LBM-Agarose Bramme sollte bei Raumtemperatur für mindestens 1 Stunde vor der Verwendung platziert werden, um sicherzustellen, dass die Agarose Bramme Raumtemperatur erreicht ist. Ort 1 ul des Phagen / Zellgemisches auf dem Agarose Bramme und warten für 1 min, damit die Mischung in die Agarose Bramme absorbieren. Sanft platzieren eine No.1 24 x 50 mm Deckglas auf der Oberseite der Bramme Agarose. Dieses Verfahren soll verhindern Abscheren der Phagen aus der infektiert Zelle (Abbildung 6).
6. Nach Zellschicksals unter dem Mikroskop
- Vorsichtig aufsetzen Deckglas auf der Bühne des Mikroskops. Für die Bildgebung, verwenden Sie ein High-Vergrößerung (zB 100x) Ziel (siehe Mikroskop-System in der Diskussion unten).
- Erwerben Sie ein Bild für den ersten Zeitrahmen gesetzt. Dieses Bild Set wird verwendet, um die ursprünglichen Zahlen und Positionen aller infizieren Phagen zu charakterisieren. Nehmen Sie eine Serie von 15 Bildern bei 200 nm z-Achse (vertikal) Intervallen. Bild Durch den YFP-Kanal. Darüber hinaus nehmen einen einzelnen in-Fokus Bild durch das Phasen-Kontrast-und mCherry Kanäle. Optimieren Sie die Lichtintensität und die Belichtungszeit, um eine ausreichende Signal zu erhalten bei gleichzeitiger Minimierung der Bleich-und Zellschäden (siehe Image Acquisition in der Diskussion unten).
- Erwerben Sie ein Zeitraffer-Film der nach der Infektion das Schicksal der Zelle. Bild der Probe in Phasen-Kontrast, YFP und mCherryKanäle in Zeitabständen von 10 min für etwa 4 Stunden. Während der Zeitraffer-Film mit einer einzigen z-Position Bild pro Kanal pro Zeitpunkt, um unnötige Belastung der Probe, die dem Bleichen und Phototoxizität führen könnten.
7. Bildanalyse
- Manuell zählen die Anzahl der Phagen und Aufzeichnung Phagen Lage und der Zellenlänge in der anfänglichen Zeitspanne. Dies kann mit einer Software wie MetaMorph oder ImageJ werden. Notieren Sie die Zellschicksale (lytische, lysogenen oder infiziert sind), Lyse Zeit und jede andere gewünschte Informationen mit dem Abspielen der Zeitraffer-Film. Um andere Zelle Schicksalen identifizieren, siehe Zeitraffer-Film in Repräsentative Ergebnisse weiter unten.
- Neben der manuellen Analyse erwähnt, können mehrere quantitative Informationen (zB Fluoreszenz-Ebene über die Zeit in einzelne Zellen) unter Verwendung von automatisierten Zell-Erkennung und Abstammungslinie Tracing-Algorithmen extrahiert werden. Wir verwenden ein Haus gebaut Matlab-Programm für tracing des Zellstammbaums und Fluoreszenz Ebenen zusammen mit dem Code für Schnitzcell Matlab Zelle Segmentierung (geschrieben von dem Elowitz am Caltech).
8. Repräsentative Ergebnisse:
Phage Plating:
Die Plaques der fluoreszenzmarkierten Phagen (in Schritt 1,6 und § 2) sind deutlich kleiner als die der Wildtyp (Abbildung 2). Wir haben daher Inkubieren Sie die Platten mindestens 12 Stunden in 37 ° C-Inkubator für die Plaques sichtbar sein.
Phage Ultrazentrifugation:
Nach Ultrazentrifugation des Phagen Probe mit dem CsCl Stufengradienten (Schritt 2.10), sollten zwei Banden sichtbar (3A). Das obere Band, an der Grenzfläche zwischen der Phagensuspension und SM / CsCl 1,3 g / ml aufweist, Zelltrümmer enthält und leeren Kapside Phagen. Das untere Band, an der Grenzfläche zwischen SM / CsCl 1,3 g / ml und 1.5 g / ml Schichten ist die Phagen-Band. This Band erscheint grünlich für das fluoreszierende Phagen λ LZ2. Die Band für Wildtyp-Phagen λ IG2903 erscheint bläulich 5. Nach Ultrazentrifugation von CsCl Gradienten Gleichgewicht in Schritt 2.12, sollte ein Phage Band sichtbar im mittleren Teil des Rohres (Abbildung 3B). Da das fluoreszierende Phagen λ LZ2 enthält eine Mischung aus gpD-EYFP und gpD Kapside, das Verhältnis von Protein-zu-DNA ist höher als die der Wildtyp. Daher ist das Band des fluoreszierenden Phagen λ LZ2 etwas hellere (scheint an einer höheren Stelle in der Röhre sein) als der Wildtyp λ IG290310.
DAPI Färbung:
Abbildung 5 zeigt eine typische Bilder nach Markierung des Phagen mit DAPI (Abschnitt 4). Die YFP und DAPI Signale eines erfolgreich gereinigt Phagen sollte nahe bei 100% Korrespondenz. Wir in der Regel feststellen, dass weniger als 1% des YFP spots enthalten keine DAPI (die Kapside ohne das virale Genom). Weniger als 1% der DAPI-Spots enthalten keine YFP (entsprechend nicht fluoreszierenden Phagen) 5.
Zeitraffer-Film:
Lytischen Zellen werden durch die Anhäufung von YFP-Fluoreszenz (grünen Kanal) innerhalb der Zelle, gefolgt durch Zelllyse erfasst. Lysogenen Zellen werden durch die Anhäufung von einheitlichen mCherry Fluoreszenz (Rot) im Inneren der Zelle und der Wiederaufnahme des normalen Zellwachstum und-teilung erfasst. Nichtinfizierten Zellen (oder Zellen, in denen Infektion ausgefallen) zeigt keine der Phänotypen oben und wird wachsen und teilen sich normal. Abbildung 7 zeigt einige Bild-Sätze von Phasen-Kontrast, YFP und mCherry Kanäle, und die entsprechenden Bilder dieser überlagerte drei Kanäle, von einem typischen Zeitraffer-Film (Abschnitt 6). Die einzelnen Phagen (grüne Flecken) sind deutlich sichtbar zu dem Anfangszeitpunkt Rahmen (Abbildung 7A). Typischerweise wird eine Anzahlvon Phagen werden auf der Zelloberfläche (vermutlich diese Zellen infizieren) gesehen, während sich andere Phagen unadsorbiert werden, wie in 7B (linke Tafel) gezeigt. Die Infektion Ergebnis wird erkennbar im Laufe der Zeit. Lytische Zyklus wird durch die intrazelluläre Produktion von neuen Phagen (grün, 7C) durch Zelllyse (Explosionsdarstellung Zellen mit Freigabe grünen Phagen, 7D) gefolgt angedeutet. Lysogenie wird durch die Produktion von mCherry vom P RE Promotor (rot, 7C) und die Wiederaufnahme der Zellwachstum und-teilung (rot, 7D) angegeben.




Abbildung 1. AB) erhalten. Die Phagen wird durch eine Reihe von Schritten (Paneele CL) gereinigt.

Abbildung 2. Phagenplaques. Plaques des fluoreszierenden Phagen (links) sind kleiner als die von Wildtyp (rechts) nach Inkubieren Platten für 12 Stunden bei 37 ° C.

Abbildung 3. Phage Bands nach Ultrazentrifugation. A) Zwei Bands sind sichtbar nach Ultrazentrifugation in einem CsCl-Stufengradienten. Der obere entspricht Zelltrümmer und leer Phagen Kapside, die untere Band enthält den gewünschten Phagen. Links: fluoreszierende Phagen, rechts:. Wildtyp B ) Eine einzelne Phagen Band ist sichtbar nach Ultrazentrifugation in einem CsCl Gleichgewicht Gradienten. Die fluoreszierende Phagen Band (links) ist grünlich, im Vergleich zu einem bläulichen Band für Wildtyp-Phagen (rechts).

Abbildung 4. Das Verfahren zur Herstellung Agarosegel Brammen.

Abbildung 5. Fluorescent Bilder von Phagen nach DAPI-Färbung. Individuelle Phagen sind leicht zu unterscheiden, und YFP und DAPI signalisiert co-lokalisieren sehr gut.

Abbildung 6. Schematische Darstellung der Phageninfektion und Imaging-Setup. Klicken Sie hier, um eine Full-Size-Version dieses Bildes zu sehen.
gure 7 "src =" / files/ftp_upload/3363/3363fig7.jpg "/>
Abbildung 7. Typische Bilder aus einem Zeitraffer-Film von Phagen-Infektion. Dargestellt sind die Phasen-Kontrast, YFP und mCherry Kanälen sowie eine Überlagerung der drei Kanäle. (A) YFP-Kanal-Bilder von der ersten Zeitrahmen. Links, wobei die Summe von YFP Bilder an verschiedenen z-Positionen. Die drei rechten Bildes sind Probe YFP Bilder an verschiedenen z-Positionen, die den verschiedenen Bereichen der Zelloberfläche. (B), (C) und (D) überlagerte Bilder (links) des Phasen-Kontrast (Mitte links), YFP (Mitte-rechts) und mCherry (rechts) Kanäle bei unterschiedlichen Zeiten. (B) Zum Zeitpunkt t = 0, zwei Zellen gesehen werden, wird jede durch einen einzigen Phagen (grüne Flecken) und eine Zelle infiziert 3 Phagen infiziert. Auch beobachtet werden einige adsorbierte Phagen. (C) Zum Zeitpunkt t = 80 min, wobei die beiden Zellen durch einzigen Phagen infiziert wurden jeweils in das lytische Stoffwechselweg weg, so anzuzeigen,d durch die intrazelluläre Produktion von neuen Phagen (grün). Die Zelle durch 3 Phagen infiziert hat in den lysogenen Stoffwechselweg, wie durch die Produktion von mCherry aus dem PRE-Promotor (rot) angedeutet worden. (D) Zum Zeitpunkt t = 2 Std. hat sich die Bahn im lytischen Zelllyse (Zelle Explosionsdarstellung) geführt, während die Zelle lysogenen § geteilt hat.
§ Left Platten Abbildung 7 (C) und (D) von Cell, 141 abgedruckt Lanying Zeng, Samuel O. Skinner, Chenghang Zong, Jean Sippy, Michael Feiss und Ido Golding, Decision Making in einem subzellulären Ebene bestimmt das Ergebnis von Bakteriophagen-Infektion, 682-691, Copyright (2010), mit freundlicher Genehmigung von Elsevier.
| Der Stamm Namen | Relevante Genotyp | Quelle / reference |
| Bakterienstämme | ||
| LE392 | supF | John Cronan, University of Illinois |
| Phage-Stämme | ||
| λ LZ1 | gpD-EYFP, cI857 Sam7 D-EYFP b :: kanR | Zeng et al. 5 |
| λ LZ2 | gpD-Mosaik, gleichen Genotyps als λ LZ1 | Zeng et al. 5 |
| Plasmide | ||
| pP RE - mCherry | mCherry unter der Kontrolle des P RE, amp R | Zeng et al. 5 |
| pPLate * D | gpD unter der Kontrolle des λ späten Promotor, amp R | Zeng et al. 5 |
Tabelle 1. Bakterienstämme,Phagen und Plasmide in dieser Arbeit verwendet.
| Dichte ρ (g / ml) | CsCl (g) | SM (ml) | Refractive Index η |
| 1,30 | 39 | 86 | 1,3625 |
| 1,50 | 67 | 82 | 1,3815 |
| 1,70 | 95 | 75 | 1,3990 |
Tabelle 3. CsCl-Lösungen in SM-Puffer (100 ml) für Stufengradienten hergestellt.
Discussion
Bakterienstämme, Phagen und Plasmide:
Stamm LE392 ist supF. Es wurde gewählt, um die Sam7 Mutation im Phagengenoms unterdrücken (siehe Tabelle 1 für weitere Details). So wird induzierten Lysogene schließlich lysieren und lassen Phagenpartikel, ebenso wie infizierte Zellen, die die lytischen Weg gewählt haben. Lysogenen Zellen werden bei 30 ° C aufgrund der Anwesenheit des temperaturempfindlichen cI 857-Allel im Phagengenom. Nach Wärmeinduktion werden gpD-EYFP und Wildtyp gpD aus dem Genom von λ LZ1 und dem Plasmid pPlate * D jeweils coexprimiert. Als Ergebnis enthält das Kapsid des neugeschaffenen Phagen λ LZ2 eine Mischung aus gpD-EYFP und gpD Proteine. Dieses Mosaik Phagen ist strukturell stabil und ausreichend Fluoreszenz auf die Detektion von einzelnen Phagen 5 zu ermöglichen. pP RE - mCherry ist ein Reporterplasmid zur Wahl des lysogenen pathwa erkenneny. Der Promotor P RE wird durch CII beim Aufbau Lysogenie 1,11 aktiviert. pP RE - mCherry 5 wurde aus pE-gfp 11 durch Austausch gfp mit mCherry 12 abgeleitet. Für weitere Details siehe unseren früheren Arbeiten 5.
Growth Zustand Parameter:
Während Lysogen Induktion (Abschnitt 1), gibt milde Schütteln bei 180 rpm ein guter Virus Ausbeute 13. Verwenden von Glucose im Wachstumsmedium sollte als Glucosemetabolismus vermieden werden erzeugt sauren Stoffwechselprodukte, und reife Lambda Partikel sind instabil bei saurem pH 13. Die Zugabe von MgSO 4 wird zur Stabilisierung des Phagen-Kapsid 3 ab. Für tragende Phagen cI Wildtyp (statt cI 857), kann die induzierte Lysogen Verwendung der DNA-schädigende Mittel Mitomycin C 3 ist. Im Schritt 1.3 wird die Inkubation bei 37 ° C normalerweise nicht länger als 90 Minuten. Es ist usef ul die Zelldichte von OD 600 Check alle 30 min. Für eine gute Lysat sinkt OD 600 auf etwa 0,2 oder weniger, und der verbleibende OD 600 ist ein Ergebnis der Zelltrümmer. Inkubation zu lange in einer niedrigeren Phagen Ertrag führen, da die neu geschaffene Phagen können beginnen, ihre DNA in Zelltrümmer zu injizieren. Um ein sichtbares Phagen Band (mindestens 1 x 10 11 Phagenpartikel) in den Schritten 2.11 und 2,13 zu erhalten, wachsen mindestens 500 ml Kultur in Schritt 1.2. Der Zusatz von 0,2% Maltose in das Wachstumsmedium in den Schritten 5.1 und 5.2 an die Induktion der Expression des LamB, der Rezeptor für den Phagen Lambda-Adsorption 3,14 abzielen. MCherry - das 1000-fache Verdünnung statt 100-fach in Schritt 5.2 wird zur Verringerung der mCherry Hintergrundpegels von der Reporterplasmid pP RE abzielen. In Schritt 5.5 für Phagen DNA-Injektion auslöst, 35 ° C gewählt wird, um die Induktion des temperatursensitiven cI857 Allel vermeiden.
Phage Reinigung:
jove_content "> Die Phagen Reinigungsschritte (Schritte 2.1 bis 2.11) können mit anderen Aufreinigungsprotokolle 5 ersetzt werden, aber die endgültige Ultrazentrifugation durch CsCl Gradienten Gleichgewicht (Schritte 2.12 und 2.13) ist unvermeidbar. Ausschwingrotoren in den Schritten 2.10 und 2.12 bis benötigten sorgen für scharfe sichtbar Phagen Bands. Erhalt einer reinen Phagen Lager kann leicht bis zu einer Woche, so ist es notwendig, die Phagentiter auf dem Weg zu überprüfen, um sicherzustellen, dass nichts schief geht während der Zwischenschritte.Phage Handhabung:
Bei allen Reinigungsverfahren in Abschnitt 2, ist es wichtig zu handhaben Phagen vorsichtig Lysat zu vermeiden Scher Phagen tails aus Phagen Köpfe. Während Zellinfektion in Abschnitt 5 (zB Schritte 5.5 bis 5.7), ist es auch wichtig, um die Scherung von Phagenpartikeln aus der infizierten Zelle zu vermeiden. Beachten Sie, dass, wenn der Phage aus der infizierten Zelle nach dem Injizieren seine DNA geschert wird, das Ergebnis ein "dunkel" Infektion ist, dh der infection Ergebnis wird im Experiment beobachtet werden, aber die Infektion Phagen nicht. Um solche Probleme zu minimieren, verwenden wir eine breite Pipettenspitze Beim Einsatz von Phagen oder Phagen / Zellgemisch.
DAPI Testing:
Färbung des Phagen Lager mit DAPI (Abschnitt 4) ist eine schnelle und effiziente Methode, um die Reinheit des Phagen Lieferverfügbarkeit überprüfen. Es kann auch verwendet werden, um mögliche Abbau eines bestehenden Phagen Lager über die Zeit zu testen. Lager für ein reines, sollte die Ko-Lokalisation von YFP und DAPI-Signale unter dem Fluoreszenzmikroskop nahe 100% liegen. Wir beobachten, dass typischerweise weniger als 1% der YFP Spots enthalten keine DAPI (repräsentiert Kapside ohne Virusgenom), was bedeutet, dass diese Teilchen nicht erfolgreich verpacken virale DNA anzeigt oder bereits ihre DNA injiziert anderswo. Weniger als 1% der DAPI-Spots enthalten keine YFP (entsprechend nicht fluoreszierenden Phagen). Ist dies nicht der Fall ist, die Schritte 2,12 bis 2,14 Notwendigkeit, in o wiederholt werdenrder wieder zu reinigen. Im Hinblick auf die Bildparameter, ist das Mikroskop Setup in Schritt 4,3 nicht so kritisch wie in Abschnitt 5, da keine langfristigen live-cell imaging hier erforderlich ist. Allerdings halten die gleichen Mikroskopie Einstellungen wie in Abschnitt 5 ist nützlich, wenn man wünscht, die Fluoreszenzintensität eines einzelnen Phagenpartikel zu kalibrieren. Falls der PBS-Agarose Bramme nicht sehr sauber oder zu sehr DAPI Farbstoff verwendet wird, können einige DAPI Stellen entsprechend Phagen-DNA mit einem "Halo" umgeben sein. Wenn zu wenig DAPI Farbstoff verwendet wird, kann das Signal von der DAPI-Kanal sehr schwach sein.
Microscope System:
Für die Bildgebung in Abschnitt 6, verwenden wir eine kommerzielle invertierten Epifluoreszenzmikroskop (Eclipse TE2000-E, Nikon) mit einer 100x-Objektiv (Plan Fluo, numerische Apertur 1,40, Öl-Immersion) und Standard-Filter-Sets (Nikon). Die Fluoreszenz Lichtquelle eine Bogenlampe mit Steuerung der Lichtintensität. Die folgenden Funktionen sind computergesteuert: x, y und z position; Hellfeld-und Fluoreszenz-Shutter und Fluoreszenz-Filter Wahl. Eine Autofokus-Funktion erforderlich ist. Andernfalls kann der Fokus leicht abdriften während der Zeitraffer-Film (in der Regel 4 Stunden lang). Die Möglichkeit, mehrere (x, y)-Positionen zu jedem Zeitpunkt zu erwerben ist nützlich, da sie mehrere Infektionsereignissen parallel verfolgen können. Wir in der Regel erwerben 8 Bühne Positionen in jedem Film, nach bis zu 100 Infektionen Veranstaltungen. Die Kamera, die wir verwenden, ist eine gekühlte 512x512 CCD mit 16x16 um Pixel-Kamera mit einem Dynamikbereich von 16 Bit (Cascade512, Photometrics). Akquisition unter Verwendung MetaMorph Software (Molecular Devices). Das Mikroskop sollte in einem temperierten Raum gestellt werden, alternativ der Mikroskoptisch sollte durch eine temperaturgesteuerte Kammer umgeben sein.
Image Acquisition:
Für Live-Cell-Imaging, ist es wichtig, um unnötige Belastung der Probe, die Bleich-und pho führen könntetotoxicity. Daher ist es am besten, zuerst zu charakterisieren Ihr System, um eine optimale Belichtung, die für die Fluoreszenz-Detektion ermöglicht zwar nicht was zu übermäßigen Bleichen oder Hemmung des Zellwachstums zu finden. Um eine gute Fluoreszenz-Bild zu erhalten, mit dem spannenden Lichtintensität, Belichtungszeit und Kamera Gewinn spielen. In den Schritten von 6,2 bis 6,3, wird das Gestell 10 min Intervall für den Zweck der Minimierung Belichtung gewählt. In jedem Rahmen ist nur ein einziger In-Fokus-Bild im Phasenkontrastmikroskop (für Zellerkennung) und fluoreszierenden Kanäle (zum Bestimmen Zellschicksals) benötigt. In dem ersten Zeitpunkt sind jedoch mehrere Bilder z-Position durch den Kanal YFP erforderlich, um alle infizierenden Phagen an der Zelloberfläche zu erfassen. Die YFP Belichtungszeit im Anfangs-Rahmen müssen gegebenenfalls auch höher sein als die für den Zeitraffer-Film in den späteren Zeitrahmen verwendet.
Image Analysis:
Sehr genau rechnen Phagenpartikel Umgebung der Zelloberfläche in Schritt 7.1. Alsbereits erwähnt, nehmen wir eine Reihe von Z-Stapel durch YFP-Kanal in Schritt 6.2. Dies kann jedoch noch verlassen einige fluoreszierende Phagenpartikel out-of-focus, die die Zählung herausfordert. Die Zellenlänge in der anfänglichen Zeitspanne wird gemessen unter Verwendung des Metamorph Software. Die Zelle kann auch durch Länge ImageJ oder andere Software-Tools zu messen. Zusätzlich kann ein automatisiertes Haus gebaut Matlab Programm sehr nützlich sein, Informationen zu erhalten, wie Fluoreszenz-Änderung über die Zeit entlang Zelllinien.
Disclosures
Keine Interessenskonflikte erklärt.
Acknowledgments
Wir sind dankbar, dass Michael Feiss und Jean Sippy für die Führung auf Phagen Erstellung und Reinigung. Wir danken Michael Elowitz für die Bereitstellung der Zell-Erkennung Software, Schnitzcell. Der Welch Foundation (Grant Q-1759) und Human Frontier Science: Arbeit in der Golding Labor ist durch Zuschüsse aus dem National Institutes of Health (R01GM082837), der National Science Foundation (Center für Physik von lebenden Zellen 082265, PFC) unterstützt Program (RGY 70/2008).
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma-Aldrich | D4527-10KU | |
| RNase | Sigma-Aldrich | R4642-10MG | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| SM buffer | TEKnova, Inc. | S0249 | |
| NZYM | TEKnova, Inc. | N2062 | |
| CsCl | Sigma-Aldrich | C3011-250G | |
| Syringe | BD Biosciences | 309585 | |
| Needle | BD Biosciences | 305176 | |
| Dialysis cassette | Thermo Fisher Scientific, Inc. | 66333 | |
| Microscope slide | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti ultra-clear tube | Beckman Coulter Inc. | 344060 | |
| SW60Ti ultra-clear tube | Beckman Coulter Inc. | 344062 | |
| SW40Ti rotor | Beckman Coulter Inc. | 331302 | |
| SW60Ti rotor | Beckman Coulter Inc. | 335649 | |
| Refractometer | Fisher Scientific | 13-947 | |
| Epifluorescence microscope | Nikon Instruments | Eclipse TE2000-E | |
| Table 2. Reagents and equipment. | |||
References
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S.
- Ptashne, M. A genetic switch : phage lambda revisited. , 3rd edn, Cold Spring Harbor Laboratory Press. (2004).
- Hendrix, R. W. Lambda II. , Cold Spring Harbor Laboratory. (1983).
- Hershey, A. D. The Bacteriophage lambda. , Cold Spring Harbor Laboratory. (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. Current protocols in molecular biology. , John Wiley & Sons. (1994).
- Sambrook, J., Russell, D. W. Molecular cloning : a laboratory manual. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Fasman, G. D. Practical handbook of biochemistry and molecular biology. , CRC press. (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Personal communication with M. Feiss. , Forthcoming.
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).

{kind=link}