

Summary
Dit artikel beschrijft de procedure voor het bereiden van een fluorescentie-gemerkte versie van bacteriofaag lambda, infectie van
Abstract
Het systeem omvat bacteriofaag (faag) en lambda de bacterie E. coli heeft lang gediend als een paradigma voor cel-lot bepaling 1,2. Na de gelijktijdige infectie van de cel door een aantal fagen, wordt een van de twee routes gekozen: lytische (virulente) of lysogene (latente) 3,4. We hebben onlangs een methode ontwikkeld fluorescerend labelen individuele fagen en konden na de infectie besluit real-time bestuderen onder de microscoop op de individuele fagen en cellen 5. We beschrijven hier de volledige procedure voor de infectie experimenten beschreven in ons eerder werk 5. Dit omvat het creëren van fluorescerende fagen, infectie van de cellen onder de microscoop imaging en gegevensanalyse. De fluorescerende faag is een "hybride" co-expressie wild-type en YFP-fusie versies van het capside-eiwit GPD. Een ruwe faag lysaat wordt eerst verkregen door het induceren van een lysogeen van de GPD-EYFP (Enhongebalanceerd Geel Fluorescent Protein) faag, drager zijn van een plasmide dat wild type GPD. Een aantal zuiveringsstappen worden vervolgens uitgevoerd, gevolgd door DAPI-etikettering en beeldvorming onder de microscoop. Dit wordt gedaan om de uniformiteit, DNA verpakking efficiëntie fluorescentiesignaal en structurele stabiliteit van de faagvoorraad verifiëren. De initiële adsorptie van fagen aan bacteriën wordt uitgevoerd op ijs, gevolgd door een korte incubatie bij 35 ° C van virale DNA injectie 6 activeren. De faag / bacteriën mengsel wordt vervolgens verplaatst naar het oppervlak van een dunne nutriënt agar plaat, bedekt met een dekglaasje en afgebeeld onder een epifluorescentiemicroscoop. De post-infectie wordt gevolgd gedurende 4 uur, 10 minuten interval. Meerdere podium posities worden bijgehouden, zodat ~ 100 cellen infecties kunnen worden opgespoord in een enkel experiment. Op elke positie en tijdstip, worden beelden die in de fase-contrast en rood en groen fluorescerend kanalen. De fase-contrast beeld wordt later gebruikt voor geautomatiseerde cell erkenning terwijl de fluorescerende kanalen worden gebruikt om de infectie resultaat karakteriseren: productie van nieuwe fluorescerende fagen (groen) gevolgd door cellysis of expressie van lysogeny factoren (red), gevolgd door weer de celgroei en celdeling. De verworven time-lapse filmpjes worden verwerkt met behulp van een combinatie van handmatige en geautomatiseerde methoden. Data-analyse resulteert in de identificatie van infectie parameters voor elke infectie gebeurtenis (bijvoorbeeld aantal en posities van fagen infecteren) als resultaat infectie (lysis / lysogeny). Aanvullende parameters kunnen worden geëxtraheerd indien gewenst.
Protocol
1. Creatie van een ruw lysaat faag (figuur 1)
- In een 50 ml kolf inoculeren een verse kolonie van LE392 (λ LZ1) [pPLate * D] (zie tabel 1 voor details) in 6 ml van LB medium 7 aangevuld met 10 ug / ml kanamycine en 100 ug / ml ampicilline. Groeien overnacht bij 30 ° C met lichte schudden (180 rpm).
- Verdun de cultuur 1:100 in LBM (LB aangevuld met 10 mM MgSO4) en groeien bij 30 ° C met lichte schudden (180 rpm). Om de faag opbrengst optimaliseren, of het kweekvolume niet meer dan een tiende van de kolf volumecapaciteit. We meestal voor te bereiden twee kolven van 2-liter of 2,5-liter, en voeg 2,5 ml overnacht cultuur in 250 ml LBM medium in elke kolf.
- Wanneer de celdichtheid OD 600 bereikt ≈ 0,6 (~ 2,5 tot 3 uur), induceren de lysogeen, door de kweek om een 42 ° C waterbad schudder gedurende 18 min met mild schudden (180 rpm) en incubate bij 37 ° C met lichte schudden (180 rpm) tot lysis zichtbaar (cultuur duidelijk, in ~ 60 - 90 min.).
- Voeg 2% chloroform aan de cultuur, schudden met de hand te mengen en incubeer 15 minuten bij kamertemperatuur. Let op: Draag handschoenen om chloroform te behandelen, en vermijd inademen.
- Breng de cultuur in twee 250 ml centrifuge flessen gecentrifugeerd cultuur in een Sorvall GSA rotor bij 10.000 rpm gedurende 15 min bij 4 ° C. Herstel de supernatant die de faagdeeltjes en gooi de pellet van vuil. Voer een tweede centrifugatie om ervoor te zorgen om zich te ontdoen van het zichtbare vuil.
- Gebruik een standaard faag titratie protocol nr. 8 bij de faag concentratie te meten. De faag titer moet ~ 5-10 x 10 9 pfu / ml. Gebruik een supF stam zoals LE392 als indicator kweek omdat de SAM7 mutatie in het genotype van de fluorescerende faag en gebruik top agar en agar platen gemaakt met rijke NZYM grotere plaques verkrijgen (Figuur 2).
2. Faag zuivering (figuur 1)
- Giet het lysaat in een grote (bijv. 2-liter) overbrengen, DNase I en RNase (1 ug / ml elk) aan het lysaat om de nucleïnezuren bevrijd van gelyseerde bacteriën verteren en incubeer 1 uur bij kamertemperatuur.
- Voeg 1 M NaCl aan het lysaat, de overdracht van de lysaat in 250 ml centrifuge flessen, en incubeer 3 uur op ijs. Centrifugeer het lysaat in een Sorvall GSA bij 10.000 rpm gedurende 15 min bij 4 ° C. Recover het supernatant. De faag titer moet gelijk zijn aan die van het ruwe lysaat, die ~ 5-10 x 10 9 pfu / ml. De toevoeging van NaCl bevordert dissociatie van faagdeeltjes uit bacteriële vuil en voor een efficiënte neerslag van faagdeeltjes door PEG 8.
- Giet het lysaat in een grote kolf, bijvoorbeeld 2-liter kolf, voeg 10% (w / v) PEG8000 in het lysaat langzaam roeren of schudden PEG8000 lossen bij kamertemperatuur. Breng de lysaat in 250 ml centrifuge flessen en incubeer overnacht (-16 uur) bij 4 ° C. Centrifugeer het lysaat in een Sorvall GSA rotor bij 10.000 rpm gedurende 15 min bij 4 ° C. Verwijder het supernatant.
- Week de pellet (faagdeeltjes geprecipiteerd met PEG8000) met faag SM buffer (4 ml SM buffer per 250 ml van initiële faag lysaat). Incuberen met een zeer milde of geen schudden schudden gedurende 16 uur bij 4 ° C.
- Verwijder voorzichtig de lysaat (SM buffer met de faagdeeltjes) in een 50 ml Eppendorf centrifugebuis en was de resterende pellet met 0,5 - 1 ml SM buffer.
- Voeg gelijk volume chloroform aan het lysaat. Meng voorzichtig het lysaat met chloroform door het omkeren op en neer voor een paar keer. Centrifugeer bij 4.000 rpm gedurende 15 min bij 4 ° C in een Eppendorf 5804R of soortgelijke tafelcentrifuge.
- Herhaal stap 2.6 tot een duidelijker lysaat te krijgen. De faag titer moet ~ 1-2 x 10 11 pfu / ml.
- Bereid SM / CsCl oplossingen met drie verschillende dichtheden (ρ) van 1,3 g / ml, 10,5 g / ml en 1,7 g / ml. Meet de brekingsindex (η) een nauwkeuriger aflezing dichtheid krijgen. De dichtheid conversie 9 is ρ = 10,8601 η - 13.4974 bij 25 ° C. Zie tabel 3 voor details.
- Gebruik een spuit met een lange naald om de oplossing te laden in een 14 ml ultraheldere Beckman ultracentrifuge 40Ti buis. Om verwisseling en een betere dichtheidsgradiënt vormen onderliggende de oplossing (dus lagen oplossingen van toenemende dichtheid onder elkaar) worden gebruikt, dwz voorzichtig 2 ml SM / CsCl oplossingen laden op bevel van 1,3 g / ml, 1,5 g / ml en 1,7 g / ml door de naald met een 3 ml spuit aan de bodem van de buis.
- Voorzichtig laden 8 ml faag lysaat door overlappen van de top van de 14 ml buis ultracentrifuge. Bereid een balans buis. Centrifugeren in een Beckman SW40Ti rotor bij 24.000 rpm gedurende 4 uur bij 4 ° C.
- Verwijder voorzichtig uit de buis in een donkere kamer en verlichten van de bovenkant van de buis tegen een zwarte achtergrond met behulp van aflashlight. De faag band moet duidelijk zichtbaar ter plaatse van het grensvlak tussen 1,3 g / ml en 1,5 g / ml SM / CsCl lagen (Figuur 3A). Punctie door de zijkant van de buis iets onder de band met een 21,5 gauge naald met een 3 ml spuit. Zachtjes verzamelen ~ 500 pi van de faagsuspensie. De faag titer moet ~ 5-10 x 10 11 pfu / ml.
- Plaats de faag suspensie in een 4 ml ultra-heldere Beekman ultracentrifuge SW60Ti rotorbuis. Vul de buis met 1,5 g / ml SM / CsCl-oplossing. Bereid een balans buis. Centrifugeren in een Beckman SW60Ti rotor bij 35.000 rpm gedurende 24 uur bij 4 ° C.
- Herhaal deze procedure als in stap 2,11 de faag verzamelen van de zichtbare band. De band moet zichtbaar zoals in figuur 3B.
- Plaats de faagsuspensie een dialysemembraan cassette (tabel 2) en dialyseer driemaal tegen een 1000-voudig volume van SM buffer bij 4 ° C gedurende perioden van 3 uur, 3 uur en overnight (~ 16 uur). Het doel van de dialyse te ontdoen van CsCl in de faagsuspensie. De uiteindelijke titer faag moet ~ 5-10 x 10 11 pfu / ml.
3. Bereid een agarose gel plaat (figuur 4)
- Schoon 6 objectglaasjes (75 x 50 mm, 1 mm dik) met 70% ethanol.
- Schik 5 glijbanen en zet hem vast met tape zoals weergegeven in figuur 4.
- Meng 0,09 g agarose in 6 ml medium in een klein bekerglas bedekt met plastic wrap (waardoor 1,5% agarose). Verhit op een hete plaat totdat de oplossing helder wordt.
- Giet de agarose oplossing op de beveiligde dia's.
- Plaats de laatste dia op de top, zorgvuldig vermijden van luchtbellen. Plaats gewicht op de top en laat het afkoelen voor ~ 30 min.
- Verwijder de 4 dia's aan de zijkant, en samen wikkel de plaat met de boven-en onderkant dia's met plastic wrap. De plaat kan worden opgeslagen bij 4 ° C gedurende 3 dagen.
4. Testen van de gezuiverde faagvoorraad
- Bereideneen PBS-agarose gel plaat zoals hierboven beschreven (hoofdstuk 3).
- Vlekken op de gezuiverde faag met DAPI. Mix 10 pi faag (~ 1 x 10 10 pfu / ml) met 10 pl van 10 ug / ml DAPI (DAPI uiteindelijke concentratie van 5 ug / ml), incubeer 30 min bij 4 ° C of 10 minuten bij kamertemperatuur.
- Place 1 ul van het faag / DAPI mengsel in het midden van een No.1 24 x 50 mm dekglaasje, overlay een klein stukje (~ 10 x 10 mm) van de bereide PBS-agarose plaat. Het stukje agarose plaat wordt gesneden met een scheermesje na de bovenste slede op de sandwich gel wordt verwijderd. Afbeelding van het monster onder de epifluorescentiemicroscoop door de YFP en DAPI kanalen. Individuele fagen moet zichtbaar buigingsbegrensde fluorescent "spots" in beide kanalen (Figuur 5). Gebruik dezelfde microscoop en camera basisinstellingen opslaan als in stap 6.2.
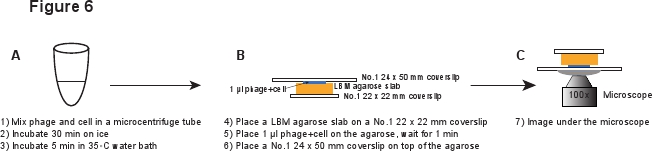
5. Infectie (Figuur 6)
- In een 14 ml Falcon buis, enten een verse kolonie van LE392 [pP RE-MCHerry] (zie tabel 1 voor details) in 2 ml LB medium aangevuld met 100 ug / ml ampicilline, 10 mM MgSO4 en 0,2% maltose. Groeien bij 37 ° C met matig schudden (265 rpm).
- Verdun de cultuur 1:1000 in LBMM (LB aangevuld met 10 mM MgSO4 en 0,2% maltose) heeft, bijvoorbeeld 5 pi overnacht cultuur in 5 ml LBMM medium in een 50 ml kolf. Groeien OD 600 ≈ 0,4 bij 37 ° C met gematigd schudden (265 rpm).
- Gebruik LBM medium een LBM-agarose gel plaat bereiden zoals beschreven in sectie 3.
- Centrifugeer 1 ml cellen bij 2.000 g in een bench-top microcentrifuge gedurende 2 minuten bij kamertemperatuur. Verwijder de bovenstaande vloeistof, en voorzichtig de cellen opnieuw in suspensie in 20 ul ijskoude LBMM aan OD 600 en 20 te bereiken.
- Bij het manipuleren van het gezuiverde faag voorraad, gebruik van een breed pipet tip of knip de reguliere pipetpunt om de tip opening breder, om te voorkomen dat het afschuiven van de faagdeeltjes 3. Voorzichtig Mix 20 pl van cellen met 20 pi gezuiverde faag een gemiddelde fagen-cel verhouding in het bereik van 0,1 te bereiken - 5. Incubeer op ijs gedurende 30 min om faag adsorptie toestaan en incuberen in een 35 ° C waterbad gedurende 5 min om faag DNA injectie 6 activeren.
- Pipetteer op en neer een paar keer naar een cel aggregaten te scheiden. Opnieuw gebruik maken van een breed pipetpunt om te voorkomen dat het afschuiven van de fagen. Verdun het mengsel 1:10 in LBMM, bijvoorbeeld 5 ul mengsel in 45 ul LBMM.
- Plaats een stuk LBM-agarose plaat (~ 10 x 10 mm) op een No.1 22 x 22 mm dekglaasje. De bereide LBM-agarose plaat moet worden geplaatst bij kamertemperatuur gedurende ten minste 1 uur vóór gebruik dat de agarose plaat kamertemperatuur. Place 1 ul van het faag / celmengsel op agarose plaat en wacht 1 min om het mengsel te absorberen in de agarose plaat. Plaats voorzichtig een No.1 24 x 50 mm dekglaasje bovenop de agarose plaat. Deze procedure is bedoeld om te voorkomen dat het afschuiven van de fagen van de infected cel (Figuur 6).
6. Na lot van de cel onder de microscoop
- Voorzichtig monteer de dekglaasje op het podium van de microscoop. Voor beeldvorming, gebruik maken van een sterk vergrotende (bijv. 100x) doelstelling (zie Microscoop System in discussie hieronder).
- Acquire een afbeelding wilt instellen voor de eerste periode. Deze afbeelding set wordt gebruikt om de initiële aantallen en posities van alle infecteren fagen karakteriseren. Een serie van 15 beelden bij 200 nm z-as (verticale) intervallen. Beeld door de YFP kanaal. Daarnaast neem een single in-focus beeld door de fase-contrast en mCherry kanalen. Optimaliseren van de lichtintensiteit en belichtingstijd om voldoende signaal te verkrijgen terwijl het minimaliseren van het bleken en celbeschadiging (zie Image Acquisition in discussie hieronder).
- Schaf een time-lapse film van de post-infectie lot van de cel. Afbeelding van het monster in fase-contrast, YFP en mCherrykanalen tijdsinterval van 10 min tot ongeveer 4 uur. Tijdens de time-lapse filmpje, gebruik dan een enkele z-positie beeld per kanaal per tijdstip, om onnodige blootstelling van het monster, wat kan leiden tot het bleken en fototoxiciteit te voorkomen.
7. Beeldanalyse
- Handmatig tellen het aantal fagen en opnemen faag locatie en de cel lengte in de eerste termijn. Dit kan gedaan worden met behulp van software zoals Metamorph of ImageJ. Noteer de celtypes (lytische, lysogene of niet-geïnfecteerde), lysis tijd, en alle andere gewenste informatie door het spelen van de time-lapse film. Om verschillende celtypes te identificeren, zie Time-lapse film in Representatieve resultaten hieronder.
- Naast de handmatige analyse bovenstaande kan meer kwantitatieve informatie (bijvoorbeeld fluorescentieniveau tijd in afzonderlijke cellen) worden geëxtraheerd met behulp van geautomatiseerde cel-herkenning en lineage tracing algoritmen. We gebruiken een huis gebouwd Matlab-programma voor tracing de celafstamming en fluorescentieniveaus samen met de Schnitzcell Matlab code voor cel segmentatie (geschreven door de Elowitz groep Caltech).
8. Representatieve resultaten:
Faag Plating:
De plaques van de fluorescent gelabelde fagen (in stap 1.6 en Deel 2) aanzienlijk kleiner dan die van wild type (figuur 2). Daarom Incubeer de platen ten minste 12 uur in 37 ° C incubator de plaques zichtbaar.
Faag Ultracentrifugatie:
Na ultracentrifugatie van de faag monster met de CsCl stapgradiënt (stap 2.10) moeten twee banden zichtbaar (Figuur 3A). De bovenste band, bij het grensvlak tussen de faagsuspensie en SM / CsCl 1,3 g / ml laag bevat celresten en lege faag capsiden. De onderste band, op het grensvlak tussen SM / CsCl 1,3 g / ml en 1,5 g / ml lagen, is de faag band. This band verschijnt groenachtig voor de tl-faag λ LZ2. De band voor wild-type faag λ IG2903 verschijnt blauwachtig 5. Na ultracentrifugatie van CsCl gradiënt evenwicht in stap 2.12, moet men faag band zichtbaar in het midden van de buis (Figuur 3B). Aangezien de fluorescerende faag λ LZ2 bevat een mengsel van GPD-EYFP en GPD capsiden de verhouding van eiwit naar DNA is hoger dan die van wild type. Daarom is de band van de fluorescerende faag λ LZ2 iets lichter (lijkt op een hogere plaats in de buis) dan wild type λ IG290310.
DAPI kleuring:
Figuur 5 toont typische beelden verkregen na het labelen van de faag met DAPI (hoofdstuk 4). De YFP en DAPI signalen van een succesvol gezuiverde faag moet bijna 100% correspondentie. We meestal zien dat minder dan 1% van de YFP spots bevatten DAPI (capsiden die zonder het virale genoom). Minder dan 1% van de DAPI spots bevatten YFP (uit niet-fluorescerende fagen) 5.
Time-lapse Movie:
Lytische cellen worden herkend door de accumulatie van YFP fluorescentie (groene kanaal) binnen de cel, gevolgd door cellysis. Lysogene cellen worden herkend door de accumulatie van uniforme mCherry fluorescentie (rood) in de cel en de hervatting van de normale celgroei en celdeling. Geïnfecteerde cellen (of cellen waar infectie heeft gefaald) geeft geen van de fenotypen boven en zullen groeien en delen normaal. Figuur 7 toont een aantal beeld-sets van fase-contrast, YFP en mCherry kanalen en de overeenkomstige overlaid beelden van deze drie kanalen, van een typische time-lapse film (hoofdstuk 6). De individuele fagen (groene vlekken) zijn duidelijk zichtbaar bij de eerste termijn (Figuur 7A). Typisch een aantalvan fagen worden gezien op het celoppervlak (vermoedelijk die cellen infecteren) terwijl andere fagen geadsorbeerde, zoals in Figuur 7B (linker paneel). De infectie uitkomst wordt onderscheiden in de tijd. De lytische cyclus wordt aangeduid door de intracellulaire productie van nieuwe fagen (groen, figuur 7C) gevolgd door cellysis (uiteengetrokken cellen model groene fagen, figuur 7D). Lysogeny wordt aangegeven door de productie van mCherry van de P RE promoter (rood, figuur 7C) en de hervatting van celgroei en deling (rood, figuur 7D).




Figuur 1. AB). De faag wordt gezuiverd door een reeks stappen (platen CL).

Figuur 2. Faagplaques. Plaques van de fluorescerende faag (links) zijn kleiner dan die van wild type (rechts) na incubatie platen gedurende 12 uur bij 37 ° C.

Figuur 3. Phage bands na ultracentrifugatie. A) Twee banden zichtbaar na ultracentrifugatie in een CsCl stapgradiënt. De bovenste komt overeen met celresten en lege faag capsiden, de onderste band bevat de gewenste faag. Links: TL-faag, rechts:. Wild-type B ) Een faag band zichtbaar is na ultracentrifugatie in een CsCl gradiënt evenwicht. De fluorescerende faag band (links) is groenachtig, in vergelijking met een blauwachtige band voor wild-type faag (rechts).

Figuur 4. De procedure voor het bereiden van agarose gel platen.

Figuur 5. Fluorescerende beelden van fagen na DAPI kleuring. Individuele fagen zijn gemakkelijk te onderscheiden, en YFP en DAPI signaleert co-lokaliseren erg goed.

Figuur 6. Schematische beschrijving van faag infectie en imaging setup. Klik hier om een grotere versie van deze foto te bekijken.
guur 7 "src =" / files/ftp_upload/3363/3363fig7.jpg "/>
Figuur 7. Typische beelden van een time-lapse filmpje van faag infectie. Getoond worden de fase-contrast, YFP en mCherry kanalen, alsmede een overlay van de drie kanalen. (A) YFP-kanaals beelden van de eerste termijn. Links de som van YFP beelden op verschillende z-posities. De drie rechter beeld sample YFP beelden op verschillende z-posities, die overeenkomen met verschillende gebieden van het celoppervlak. (B), (C) en (D) Overlaid beelden (links) van de fase-contrast (midden-links), YFP (midden-rechts) en mCherry (rechts) kanalen op verschillende tijdspaden. (B) op t = 0, twee cellen worden gezien wordt elke geïnfecteerd door een faag (groene vlekken) en een cel die is geïnfecteerd met 3 fagen. Ook waargenomen sommige geadsorbeerde fagen. (C) op t = 80 min, de twee cellen geïnfecteerd bij een enkele fagen hebben elk gegaan in de lytische route, zoals aangegevend de intracellulaire productie van nieuwe fagen (groen). De cel geïnfecteerd met 3 fagen gestoken in de lysogene route, zoals aangegeven door de productie van mCherry van de PRE promoter (rood). (D) Op t = 2 uur, werd de lytische route resulteerde in lysis (cel uiteengetrokken), terwijl de lysogene cel verdeeld §.
§ Linker panelen van Figuur 7 (C) en (D) worden herdrukt van Cell, 141, Lanying Zeng, Samuel O. Skinner, Chenghang Zong, Jean Sippy, Michael Feiss, en Ido Golding, de besluitvorming op een subcellulaire niveau bepaalt de uitkomst van Bacteriofaag Infectie, zeshonderdtweeentachtig-zeshonderdeenennegentig, Copyright (2010), met toestemming van Elsevier.
| Stamnaam | Relevante genotype | Bron / referentie |
| Bacteriestammen | ||
| LE392 | supF | John Cronan, Universiteit van Illinois |
| Faagstammen | ||
| λ LZ1 | GPD-EYFP, cI857 SAM7 D-EYFP b :: kanR | Zeng et al.. 5 |
| λ LZ2 | GPD-mozaïek, hetzelfde genotype als λ LZ1 | Zeng et al.. 5 |
| Plasmiden | ||
| pP RE - mCherry | mCherry onder controle van P RE, ampR | Zeng et al.. 5 |
| pPLate * D | GPD onder de controle van λ late promoter, versterker R | Zeng et al.. 5 |
Tabel 1. Bacteriestammen,fagen en plasmiden gebruikt in dit werk.
| Dichtheid ρ (g / ml) | CsCl (g) | SM (ml) | Brekingsindex η |
| 1,30 | 39 | 86 | 1,3625 |
| 1,50 | 67 | 82 | 1,3815 |
| 1,70 | 95 | 75 | 1,3990 |
Tabel 3. CsCl bereide SM buffer (100 ml) voor stap verlopen.
Discussion
Bacteriestammen, plasmiden en faag:
Strain LE392 is supF. Werd gekozen om de SAM7 mutatie onderdrukken in het faag genoom (zie tabel 1 voor details). Aldus zullen geïnduceerde lysogenen uiteindelijk lyseren en laat faagdeeltjes, evenals geïnfecteerde cellen die gekozen lytische route. Lysogene cellen worden gekweekt bij 30 ° C door de aanwezigheid van het temperatuurgevoelige cI 857 allel in het faaggenoom. Na hitte-geïnduceerde worden GPD-EYFP en wildtype GPD co-expressie uit het genoom van λ LZ1 en het plasmide pPlate * D respectievelijk. Daardoor het capside van het nieuwe faag λ LZ2 bevat een mengsel van GPD-EYFP en gpd eiwitten. Dit mozaïek faag is structureel stabiel en voldoende fluorescerende om detectie van individuele fagen 5 mogelijk te maken. pP RE - mCherry is een reporter plasmide gebruikt om de keuze van de lysogene pathwa detectereny. De promotor P RE wordt geactiveerd door CII tijdens de oprichting van lysogeny 1,11. pP RE - mCherry 5 werd afgeleid van PE-GFP 11 door het vervangen van GFP met mCherry 12. Voor meer details zie onze eerdere werk 5.
Groei Conditie Parameters:
Tijdens lysogeen inductie (afdeling 1), milde schudden bij 180 rpm geeft een goed virus opbrengst 13. Gebruik van glucose in het groeimedium worden vermeden glucosemetabolisme genereerd zure metabolieten en volwassen lambda deeltjes niet stabiel bij zure pH 13. De toevoeging van MgSO4 is gericht op het stabiliseren van de faag capside 3. Voor fagen die wild type Ci (in plaats van cI 857), kan het lysogeen worden geïnduceerd met behulp van de DNA-beschadigende middel mitomycine C 3. In stap 1,3, de incubatie bij 37 ° C normaliter niet langer dan 90 minuten. Het is USEF ul de celdichtheid controleren door OD600 elke 30 min. Voor een goede lysaat OD 600 daalt tot ongeveer 0,2 of minder, en de resterende OD 600 is een gevolg van celresten. Incuberen te lang kan leiden tot een lagere opbrengst faag aangezien de nieuwe faag kan beginnen hun DNA te injecteren in celresten. Als u een zichtbare faag band (minimaal 1 x 10 11 faagdeeltjes) te verkrijgen in de stappen 2,11 en 2,13, groeien ten minste 500 ml cultuur in stap 1.2. De toevoeging van 0,2% maltose in het groeimedium in stappen 5.1 en 5.2 is gericht op het induceren van expressie van LamB, de receptor voor faag lambda adsorptie 3,14. De 1000-voudige verdunning in plaats van 100-voudig in stap 5.2 is gericht op het verminderen van de mCherry achtergrondniveau van de reporter plasmide pP RE - mCherry. In stap 5.5 voor faag DNA injectie triggering, 35 ° C wordt gekozen om inductie van de temperatuurgevoelige cl857 allel voorkomen.
Phage Zuivering:
jove_content "> De faag zuiveringsstappen (Stappen 2.1 tot 2.11) kan worden vervangen door andere zuivering protocollen 5, maar de uiteindelijke ultracentrifugatie door CsCl gradiënt evenwicht (stappen 2.12 en 2.13) onvermijdelijk. Swinging bucket rotors nodig in stappen 2.10 en 2.12 tot zorgen voor scherpe zichtbaar faag bands. Het verkrijgen van een zuivere faag voorraad kan gemakkelijk tot een week duren, dus het is noodzakelijk om de faag titer controleren langs de manier om ervoor te zorgen dat er niets mis gaat tijdens de tussenliggende stappen.Faag Handling:
Tijdens alle zuivering procedures van afdeling 2, is het essentieel om te behandelen faag voorzichtig lysaat om te voorkomen dat afschuiven faag staarten uit faag hoofden. Tijdens cel infectie deel 5 (bijvoorbeeld stappen 5.5 tot 5.7), is ook cruciaal voor het scheren van faagdeeltjes van de geïnfecteerde cel te voorkomen. Merk op dat als de faag wordt geschoren van de geïnfecteerde cel na injecteren zijn DNA, is het resultaat een "donker" infectie, dat wil zeggen de inFection uitkomst zal worden waargenomen in experiment, maar het infecterende fagen niet. Om dergelijke problemen te minimaliseren, gebruiken we een breed pipetpunt wanneer het hanteren fagen of de faag / cel mengsel.
DAPI Testen:
Kleuren van de faag voorraad met DAPI (hoofdstuk 4) is een snelle en efficiënte methode om de zuiverheid van de faag voorraad te controleren. Het kan ook worden gebruikt voor het testen van mogelijke afbraak bestaande faagvoorraad tijd. Voor een zuivere beelden moet de co-lokalisatie van YFP en DAPI signalen onder de fluorescentiemicroscoop dicht bij 100%. We merken op dat kenmerkend minder dan 1% van de YFP spots niet DAPI (capsiden die zonder het virale genoom), wat aangeeft dat deze deeltjes niet succesvol verpakken van het virale DNA of al hun DNA geïnjecteerd elders bevatten. Minder dan 1% van de DAPI spots bevatten YFP (uit niet-fluorescerende fagen). Indien dit niet het geval is, stappen 2,12 tot 2,14 moeten worden herhaald in oestellen opnieuw zuiveren. Met betrekking tot imaging parameters, de microscoop setup in Stap 4.3 is niet zo belangrijk als in deel 5 omdat geen langdurige live-cell imaging is hier vereist. Maar door dezelfde microscopie instellingen als in punt 5 is handig als men het ijken van de fluorescentie-intensiteit van een faagdeeltje. Als de PBS-agarose plaat is niet erg schoon of teveel DAPI kleurstof wordt gebruikt, kunnen sommige overeenkomen met DAPI spots faag DNA zijn omringd door een "halo". Indien te weinig DAPI kleurstof wordt gebruikt, kan het signaal van het kanaal DAPI zeer zwak.
Microscoopsysteem:
Voor de beeldvorming in hoofdstuk 6, gebruiken we een commercieel omgekeerde epifluorescentiemicroscoop (Eclipse TE2000-E, Nikon) met een 100x objectief (Plan Fluo, numerieke apertuur 1.40, olie-immersie) en standaard filtersets (Nikon). De fluorescentie lichtbron is een Booglamp met controle van de lichtintensiteit. De volgende functies zijn computer gestuurde: x, y en z position, helderveld en fluorescentie luiken, en fluorescentie filter keuze. Een auto-focus functie is vereist. Anders kan de focus gemakkelijk weg te drijven tijdens de time-lapse filmpje (normaal 4 uur lang). De mogelijkheid om meerdere (x, y) posities te verwerven op elk tijdstip is nuttig omdat het laat toe om meerdere infecties gebeurtenissen in parallel. We meestal verwerven 8 stadium posities in elke film, na maximaal 100 infectie gebeurtenissen. De camera die we gebruiken is een gekoelde 512x512 CCD met 16x16 micrometer pixel camera met een dynamisch bereik van 16 bits (Cascade512, Photometrics). Verwerving uitgevoerd met Metamorph software (Molecular Devices). De microscoop moet worden geplaatst in een temperatuur-geregelde kamer, als alternatief, de microscooptafel worden omringd door een temperatuur-geregelde kamer.
Image Acquisition:
Voor live-cell imaging, is het essentieel om te voorkomen dat onnodige blootstelling van het monster, wat kan leiden tot het bleken en phototoxicity. Daarom is het best eerst karakteriseren systeem een optimale belichting waardoor voor fluorescentiedetectie zonder dat leidt tot overmatige bleken of remmen van celgroei vinden. Om een goede fluorescentie beeld te verkrijgen, spelen met de spannende lichtintensiteit, belichtingstijd en de camera te krijgen. In stappen 6.2-6.3, wordt de 10 min rasterinterval gekozen met het oog op het minimaliseren van blootstelling aan licht. In elk frame wordt slechts een in-focus beeld nodig fasecontrast (voor celherkenning) en fluorescent kanalen (voor de bepaling lot van de cel). In het eerste tijdstip, echter, multiple z-positie beelden via de YFP kanaal nodig om alle infecteren fagen op het celoppervlak te vangen. De YFP belichtingstijd in het eerste frame kan ook hoger moeten zijn dan die wordt gebruikt voor de time-lapse filmpje in de latere termijnen.
Beeldanalyse:
Heel voorzichtig tellen faagdeeltjes in het celoppervlak in stap 7.1. Alshierboven is opgemerkt, nemen we een reeks van z-stacks door middel van YFP kanaal in stap 6.2. Dit kan echter nog steeds laat sommige tl-faagdeeltjes out-of-focus, die het tellen uitdagingen. De cel lengte in de eerste tijd wordt gemeten met behulp van de Metamorph software. De cel lengte kan ook worden gemeten door ImageJ of andere software. Bovendien kan een geautomatiseerd huis gebouwd Matlab-programma zeer nuttig zijn bij het verkrijgen van informatie, zoals fluorescentie verandering in de tijd langs cellijnen.
Disclosures
Geen belangenconflicten verklaard.
Acknowledgments
We zijn dankbaar voor Michael Feiss en Jean Sippy voor de begeleiding op de faag creatie en zuivering. Wij danken Michael Elowitz voor het verstrekken van de cel herkenning software, Schnitzcell. Werk in de Golding lab wordt ondersteund door subsidies van de National Institutes of Health (R01GM082837), de National Science Foundation (082.265, PFC: Centrum voor de fysica van levende cellen), de Welch Foundation (Grant Q-1759) en Human Frontier Science Programma (RGY 70/2008).
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma-Aldrich | D4527-10KU | |
| RNase | Sigma-Aldrich | R4642-10MG | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| SM buffer | TEKnova, Inc. | S0249 | |
| NZYM | TEKnova, Inc. | N2062 | |
| CsCl | Sigma-Aldrich | C3011-250G | |
| Syringe | BD Biosciences | 309585 | |
| Needle | BD Biosciences | 305176 | |
| Dialysis cassette | Thermo Fisher Scientific, Inc. | 66333 | |
| Microscope slide | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti ultra-clear tube | Beckman Coulter Inc. | 344060 | |
| SW60Ti ultra-clear tube | Beckman Coulter Inc. | 344062 | |
| SW40Ti rotor | Beckman Coulter Inc. | 331302 | |
| SW60Ti rotor | Beckman Coulter Inc. | 335649 | |
| Refractometer | Fisher Scientific | 13-947 | |
| Epifluorescence microscope | Nikon Instruments | Eclipse TE2000-E | |
| Table 2. Reagents and equipment. | |||
References
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S. Switches in bacteriophage lambda development. Annu. Rev. Genet. 39, 409-429 (2005).
- Ptashne, M. A genetic switch : phage lambda revisited. , 3rd edn, Cold Spring Harbor Laboratory Press. (2004).
- Hendrix, R. W. Lambda II. , Cold Spring Harbor Laboratory. (1983).
- Hershey, A. D. The Bacteriophage lambda. , Cold Spring Harbor Laboratory. (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. Current protocols in molecular biology. , John Wiley & Sons. (1994).
- Sambrook, J., Russell, D. W. Molecular cloning : a laboratory manual. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Fasman, G. D. Practical handbook of biochemistry and molecular biology. , CRC press. (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Personal communication with M. Feiss. , Forthcoming.
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).

{kind=link}