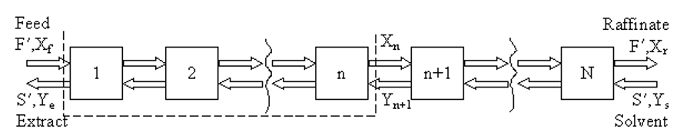
Either the a) McCabe-Thiele method, or b) a process simulator (e.g., ASPEN Plus, HYSYS, ChemSep) may be used to estimate the number of equilibrium (theoretical) stages. The McCabe-Thiele method is employed on a solvent-free basis, meaning both the solubilities of the solvent in the extract and of the diluent compound in the raffinate are neglected. A stagewise representation of counter-current liquid-liquid extraction is shown in Figure 1, where F' is the molar flow rate of the feed (approximately constant), S' is the molar flow rate of extract (approximately constant), Xf is the mole fraction of solute in feed, Ys is mole fraction of solute in the solvent, Ye is mole fraction of solute in solvent in extract stream, and Xr is mole fraction of solute in diluent in the raffinate stream.
Figure 1: Stagewise representation of the extraction process.
At steady state, a material balance on the solute between the feed end of the column and any stage, n (dotted outline above) leads to the operating line:
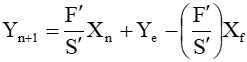 (1)
(1)
In particular, the equation is satisfied at both ends of the column, so the points (Xf, Ye) and (Xr, Ys) lie on the line.The equilibrium data in the Appendix can be used in conjunction with this equation (either graphically or numerically) to step through the column.
The process simulators can do more rigorous stage-to-stage calculations, but still assuming equilibrium stages. Either the NRTL or UNIQUAC methods (both sets of parameters in the Appendix) can be used to model the equilibrium relationship. Note that the big advantage of the simulators is that they DO tell you how much solvent winds up in the extract and how much diluent winds up in the raffinate. They also can give the exit temperatures for an adiabatic column, or the heat duty needed to keep the column isothermal.
A York-Scheibel apparatus is shown in Figure 2. Feed can be introduced at the bottom (11 stages) or at the middle of the column (6 stages).
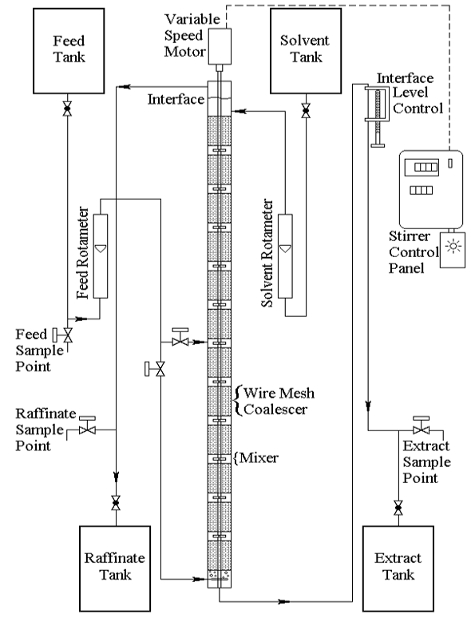
Figure 2: York-Scheibel liquid-liquid extraction apparatus.
The extraction unit consists of a 2" I.D. Pyrex column, with 11 extraction stages, each consisting of a one-inch mixing section and a four-inch wire mesh packing (coalescing) section. The column is mechanically agitated by paddlewheel-type (Rushton turbine) agitators. A variable speed motor, with a control knob and digital readout on the control panel, controls the speed of the agitator. Rotameters on the feed and solvent inlets are used to measure those flow rates. Flow rates of the extract and raffinate can be measured with a graduated cylinder and stopwatch.
The following equations relate the rotameter readings to volumetric flow rates (the flows can also be checked with a graduated cylinder):
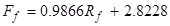 (2)
(2)
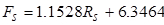 (3)
(3)
where Ff is the feed flow rate (~10 wt. % IPA) in mL/min,Rf is the feed rotameter reading,Fs is the solvent flow rate in ml/min, andRs is the solvent rotameter reading.
Figures 3 and 4 show results when both the agitation and feed flow rates were varied over a wide range. The overall efficiency and recovery increase before becoming asymptotic, which is fairly typical of liquid-liquid extractors that are not at or near flooding. At near flooding conditions, the overall efficiency and recovery are expected to sharply decrease. Note that, unlike distillation, flooding can take place in liquid-liquid extraction at either high solvent or high feed rates (or ratios).1 In this experiment, the lighter organic phase is also the dispersed (droplet) phase, so at high feed rates it is expected that the droplets coalescence prior to flooding, leading to lower rates of mass transfer and, therefore, lower recoveries and efficiencies. At high solvent rates the droplets should remain small, so it is expected that the recovery and efficiency remain high until very near the flooding point.
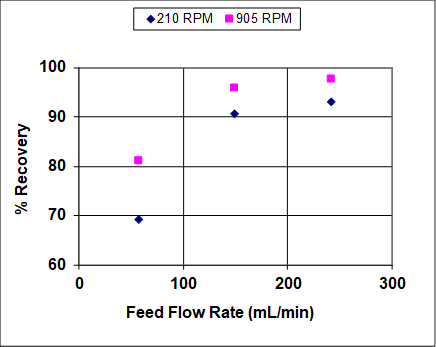
Figure 3: Percent recovery of IPA from hydrocarbon mixture into water, for a York-Scheibel column, 11 stages, 16 – 18 mol% IPA in Isopar E (feed), S/F (molar) = 1.5.
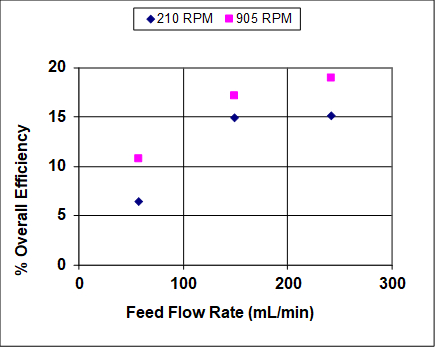
Figure 4: Percent overall stage efficiency for IPA extraction using a York-Scheibel column, 11 stages, 16 – 18 mol% IPA in hydrocarbon mixture (feed), S/F (molar) = 1.5.
As seen from Figures 3 and 4, increasing the agitation rate increases both the recovery and overall efficiency. This is because with greater power input the droplets of the dispersed phase are smaller – the observed dependence is roughly inverse with respect to agitator speed.4 The "a" parameter (interfacial area/total volume) that appears in mass transfer correlations and fundamental mass flux equations can be written as follows for uniform-size spherical droplets:
a = 6 ε/d (4)
where ε is the volume fraction of the dispersed phase. While ε can increase with an increase in either phase's superficial velocity, its changes are usually less marked than the change of diameter with respect to speed. So (usually) the more speed, the more interfacial area, leading to faster mass transfer.
The exception to the above discussion is at very high speeds, which were not reached in Figs. 3 and 4, where the two phases are so well-mixed that if the interfacial tension between them is low, emulsification will take place. The formation of an emulsion negatively impacts recovery and efficiency because the phases can no longer separate cleanly to move up or down to the next physical stage.Emulsification is a problem in many liquid-liquid extractions and where it cannot be limited a series of mixer and settler vessels in series is often preferred to column-type designs such as sieve trays or York-Scheibel units.
Liquid-liquid extraction (LLE) is an alternative to distillation which relies upon solvent-feed immiscibility (or slight miscibility) and favorable solute partition coefficients to attain high solute recoveries in a solvent phase at as low a solvent/feed ratio as practical. Although the range of flows (the "turndown") over which LLE will be effective is often limited, and while stage efficiencies are low such that phase equilibrium is not attained, certain mixtures just cannot be separated using other methods in a continuous countercurrent process. Mathematical analysis of the equilibrium operation of such extractors follows a familiar McCabe-Thiele-type procedure (although reflux is often lacking, so only one operating line). The non-equilibrium ("rate-based") analysis of LLEs is complex and depends strongly on the relative velocity between the two phases (the "slip velocity"), bubble size, and dispersed phase fraction, all of which can be observed but are difficult to predict.
To perfectly describe the hydraulics and mass transfer of a typical LLE is beyond the capability of even the most sophisticated process simulators, at present. Therefore, design of industrial units still relies on scale-up from pilot-plant-type units, such as that which was tested in this experiment. Normally the engineer attempts to duplicate key descriptors such as the "a" parameter, solvent/feed ratio, total agitator power input/volume, feed location and number of physical stages to keep the stage efficiency and recovery constant during scale-up. Even so, scale-up is an inexact science, and impurities, which alter the interfacial tension, can greatly impact the performance of real systems. The more factors that are held constant, the more likely the scale-up will be successful.
There are many different LLE contactors: a series of mixers – settler vessels, structured packings similar to those used in absorbers and distillation columns, sieve tray columns, rotating disk contactors (similar to the York-Scheibel, but with baffles instead of mesh), Kuhni contactors (a combination of rotating disk and sieve trays), and Podbielniak contactors ("Pods"), where the flow is radial and centrifugal force is used to enhance liquid phase separation.5
A classic example of industrial LLE is the separation of acetic acid from water using ethyl ether or ethyl acetate;6 it is preferred over distillation at lower acetic acid concentrations. Possibly the biggest volume LLE process is that of propane deasphalting, which is used to refine lubricating oils in refineries at near-supercritical conditions.1 However, most applications are found in the production of specialty chemicals and in pharmaceutical industries, ranging from citric acid extraction from fermentation broth to purification of antibiotics and protein purifications.1In these cases, a wide variety of oxygenated organic solvents or two-phase aqueous systems (with one phase being mostly water and the other aqueous dissolved salts and polymers) are utilized. For the latter, a typical polymer system is poly(ethylene glycol)/dextran with NaCl and Na2SO4 as salts. Applications include red blood cell separation and extraction of the phophofructokinase enzyme from S. cerevisiae.7
Appendix – Equilibrium Data8
Experimental Tie Lines in Mole Percent at 25 °C

Specific Model Parameters in Kelvin
| UNIQUAC | NRTL (a = 0.2) | ||||
| I | J | AIJ | AJI | AIJ | AJI |
| 1 | 2 | -186.05 | 104.6 | 814.26 | -468.11 |
| 1 | 3 | 361.91 | 621.82 | 3151 | 1367.4 |
| 2 | 3 | -126.43 | 311.7 | 581.79 | -25.91 |
R1 = 0.92 R2 = 2.7792 R3 = 6.523
Q1 = 1.4 Q2 = 2.508 Q3 = 5.476
| Mean Deviation between Calculate and Experimental Concentrations in Mol. % | |
| UNIQUAC (specific parameters) | 1.4 |
| NRTL (specific parameters) | 0.54 |
| UNIQUAC (default parameters) | 1.68 |