



Overview
Source: Susannah C. Shissler1, Tonya J. Webb1
1 Department of Microbiology and Immunology, University of Maryland, Baltimore, MD 21201
Immunoprecipitation (IP, also known as a 'pull-down' assay) is a widely used technique that has applications in a variety of fields. First conceived in 1984, it was refined in 1988 (1, 2). The fundamental goal of IP is purification and isolation of a specific protein using an antibody against that protein. The word "immuno" refers to the use of an antibody while the word "precipitation" refers to pulling down a specific substance from a solution. The target protein might be endogenous or recombinant. Most recombinant proteins have an epitope tag (i.e. myc or flag) attached to them to simplify subsequent purification. Typically, it is easier to optimize recombinant protein IP because the antibodies against recombinant epitope tags are very strong and effective. Antibodies against endogenous proteins have extremely variable efficacy - making it much more difficult to optimize these IPs. A necessary step after immunoprecipitation is verification of purification. The isolated protein is resolved using SDS-PAGE and subsequently probed for purity by western blots (Figure 1). An important control is the use of a different antibody during the Western blot to verify pull down of the correct protein. The combination of IP with subsequent techniques is a powerful analysis tool. The goal after purification may be characterization of the protein itself by NMR, mass spectrometry, and in vitro assays, or analysis of the protein's interacting partners (i.e. protein, DNA, RNA) (3, 4, 5).
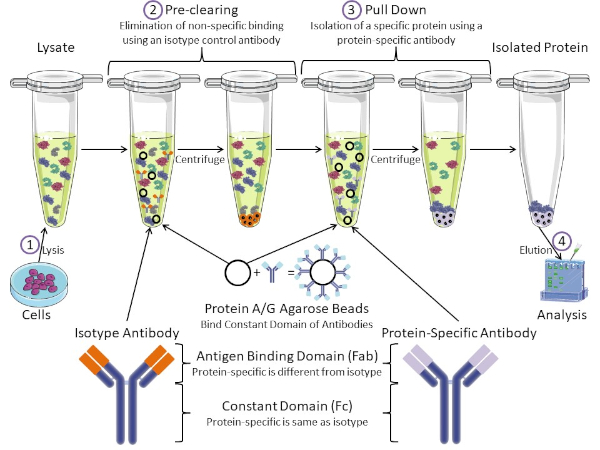
Figure 1: Overview of Immunoprecipitation Procedure. Immunoprecipitation is the isolation of a specific protein using an antibody. After production of lysate from cells, there are two major steps- pre-clearing and pull down. During pre-clearing step, the cell-lysates are pre-cleared of proteins that bind to antibodies non-specifically using an isotype control antibody. In pull down step, the target protein is pulled down using a protein-specific antibody. The isolated protein is then analyzed by Western blot. Isotype antibodies and protein specific antibodies have the same constant domain, but different antigen binding domains. A key component of this protocol is Protein A/G agarose beads that bind the constant domain of antibodies- allowing immunoprecipitation of the target protein. Please click here to view a larger version of this figure.
Antibodies are the key component of an immunoprecipitation that differentiate it from other forms of protein purification (i.e. nickel affinity column purification). Antibodies are molecules made by B cells that can recognize specific protein epitopes. Antibodies have two domains: constant (Fc) and antigen binding (Fab) (Figure 1). The constant domain identifies the type of antibody and dictates function in vivo. Usually, the constant domains of antibodies used for IP are mouse, rat, or rabbit IgG. The antigen binding portion of the antibody recognizes a specific epitope of a specific protein. Antibodies can recognize epitopes on folded proteins that may not exist when the protein is denatured and vice versa. Therefore, the availability of the epitope depends on protein folding - identifying an important factor to consider when choosing antibodies and conditions for IP.
Both prokaryotic and eukaryotic systems have antibody-binding proteins. In eukaryotic systems, the purpose is immune protection from bacteria while in prokaryotic systems, the purpose is protection from the immune system. Antibody-binding proteins affect IP methodology in two ways. First, there is a necessary pre-clearing step (Figure 1) to rid the lysate of proteins that bind antibodies - thereby reducing non-specific binding in the final product. This step uses an isotype antibody that has the same constant domain as but a different antibody binding domain than your protein-specific antibody. Bacterial antibody-binding proteins are the second key component of this method. After the protein-specific antibody binds the target protein, the antibody: protein complex must be pulled down (Figure 1). Proteins A, G, and L are bacterial proteins that bind the constant domain of antibodies. While bacteria use this to subvert the immune system, researchers have co-opted this system for easy antibody purification, and it is used during both the pre-clearing and pull-down steps. These proteins have different binding affinities for different species and different constant domain subtypes - another factor to consider when choosing conditions for IP. Many companies sell Protein A/G labeled agarose beads (Figure 1), pre-made spin columns, or resins to make columns. In general, beads and spin columns are used for smaller sample sizes while resins are used for bulk purification.
In this lab exercise, we demonstrate how to purify the endogenous protein c-myc, from primary murine thymocytes, using Protein A/G Plus agarose beads based basic immunoprecipitation technique. The protocol starts from cell lysate preparation and ends with the with verification of successful protein pull down using Western blot analysis.
Procedure
1. Immunoprecipitation using Protein A/G PLUS Agarose Beads
Cell Lysate Preparation
- Centrifuge 108 thymocytes in a microcentrifuge at 13,000 rpm for 3 min and remove the supernatant.
Note: The cell number will vary depending on the expression levels of the desired protein and the chosen cell type. - Re-suspend the cells in 500 µL lysis buffer RIPA with PMSF.
- Disrupt cells using a few quick pulses with a vortex and then aspirate the lysate a few times with a 25 G needle attached to a syringe.
Note: Avoid creating bubbles. Use a larger needle such as a 21G needle for larger cell types. - Incubate the cell lysate on ice for 10 min.
- Centrifuge the lysate at 13,000 rpm for 15 min at 4°C.
- Transfer the supernatant to a fresh, labeled microcentrifuge tube.
Pre-clearing
- Add 20 µL Protein A/G PLUS agarose beads and 1 µg of an isotype control antibody (here, mouse IgG1 isotype control antibody is used), to the lysate.
Note: The choice of isotype antibody used will depend on the protein-specific antibody used in later in the pull-down step. - Incubate the lysate mix on a centrifuge rotator in the cold room (4°C) for 30 min.
- Centrifuge the sample at 3200 rpm for 30 s at 4°C.
- Transfer the pre-cleared supernatant to a fresh, labeled, 1.5 mL microcentrifuge tube. Discard the pellet.
Protein Concentration Determination
- Determine the protein concentration of the cell lysate by performing a Bradford Assay.
- Aliquot 1000 µL Bradford Reagent into 7 microcentrifuge tubes.
- Add the following amounts of BSA protein standard (2 mg/mL) into 6 of the tubes (Table 1).
| Tube Number | BSA Volume (μL) (2 mg/mL) | Protein Concentration (μg/μL) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
Table 1: BSA protein standard amounts
- In the 7th tube, add 1 µL of the pre-cleared lysate.
Note: To ensure that the sample concentration lies within the assay detection range, prepare and analyze a 1:2 or 1:5 lysate dilution as well. - Place 200 µL from each of the 7 tubes into individual wells of a flat-bottom, 96-well plate repeating each sample in triplicate.
- Read plate on a plate reader at 595 nm.
- Generate the standard curve in excel and calculate the protein concentration of the pre-cleared lysate.
Pull down
- Label two fresh 1.5 mL microcentrifuge tubes- one as 'control' and other as 'test,' which in this example is c-myc.
- Place 500 µg of pre-cleared lysate into each of these tubes.
Note: The amount of protein used here will depend on the amount of the protein desired to be purified. - Bring up the total volume for each tube up to 500 µL using lysis buffer.
- Add 2 µg of anti-c-myc antibody to the test group tube and 2 µg mouse IgG1 isotype control antibody to the control group.
Note: Quantity of antibody will depend upon antibody efficacy and the amount of the target protein. - Incubate the tubes on a rotator in the cold room (4°C) for 2 h.
- Add 20 µL Protein A/G PLUS agarose beads to each tube.
Note: It is advisable to use pipette tips with the end cut off to prevent damage to the beads. - Incubate on a rotator in cold room (4°C) overnight.
Note: Depending on the target protein and the antibody efficacy, this step can vary from 1 h to overnight. - Centrifuge the tubes at 3200 rpm for at 30 s 4°C to pull down the beads.
- Aspirate the supernatant from each tube.
Note: The target protein is now bound to the beads. - Wash the beads two times using 500 µL 1X Dulbecco's PBS.
- Centrifuge the tubes at 3200 rpm for at 30 s 4°C.
Note: For more stringent washing, use more stringent buffers, such as RIPA. - Aspirate the buffer from each tube. Using gel-loading tips, remove any leftover buffer from beads and keep the beads on ice to elute the protein.
Note: In this example, the protein is eluted into SDS-PAGE running buffer by boiling the beads, for Western blot analysis. This approach is suitable for verifying IP results or to examine protein-protein interactions. For other downstream applications, such as purification of proteins for structural or enzymatic analysis, more sophisticated systems, such as epitope tags (flag-tag or myc-tag) are used to avoid elution of the antibody with the protein of interest.
2. IP Verification Through Western Blot Analysis
SDS-PAGE Electrophoresis:
- Re-suspend beads in 20 µL SDS-PAGE loading dye containing β-mercapto-ethonol.
- Boil the samples at 95 °C for 5 min.
- Centrifuge the beads at 13,000 rpm for 10 s at room temperature.
- Using gel loading tips, carefully pipet the samples obtained from the beads and load them into wells of 4-15% gradient SDS-PAGE gel.
- In addition to the samples, load a lane with a protein ladder as well as a lane with the pre-cleared lysate to serve as a loading control.
- Run at 100 V until dye front reaches the bottom of the gel (~1h).
Western Blot Analysis:
- Make Western blot sandwich, ensuring that PVDF membrane is between gel and red cathode.
- Transfer for 1 h at 100 V.
- Place the membrane in 5 mL blocking buffer at room temperature for 1 h on a rocker at a low setting, to block the non-specific protein binding sites.
Note: The volumes of blocking buffer, primary antibody, secondary antibody, and washes may need to be increased for larger sized blots. - Incubate the blot with 5 mL anti-c-myc antibody in the blocking buffer overnight at 4°C on rocker at a low setting.
Note: The antibody used here should be different than the one used in the pull-down step. - Wash the blot 3-6 times using 5 mL TBST with each wash being 5 min at room temperature on a rocker at a low setting.
- Incubate the blot with HRP-tagged anti-rabbit light chain secondary antibody in blocking buffer, for 1 h at room temperature on rocker at a low setting.
Note: The choice of secondary antibody will depend on primary antibody used for the Western blot. Additionally, a light chain specific secondary is used in the protocol because the target protein is close in molecular weight to the heavy chain of the antibody. If the target protein is close to 50kDa, use a light chain specific secondary. If the target protein is close of 25kDa, use and heavy chain specific secondary. - Wash the blot 3-6 times using 5 mL TBST with each wash being 5 min at room temperature on a rocker at a low setting.
- Remove liquid from blot and dab edge of blot on laboratory wipes to remove excess liquid.
- Cover blot with 1x chemiluminescent detection reagent' and incubate for 1 min.
Note: The following steps should be done in quick succession as the detection reagent is light and time sensitive. - Dab edge of blot on laboratory wipes to remove excess detection reagent.
- Place the blot on imaging surface of Imager tray.
Note: Chemiluminescent blots can also be visualized using film. - Image using the 'Chemiluminescent Program' to capture multiple time points from 10 s to 5 min.
Note: The optimal time may change based on protein quantity and the quality of antibody. - Chose an image with optimal band visibility and then export that image.
- Prior to moving the blot, take a picture of the blot using the imager, to capture the location of the ladder. Then, export that image as well.
- Using a slide preparation software (such as PowerPoint), align the bands and ladder images to form a single image.
Immunoprecipitation, or IP, is a widely used technique to isolate a protein of interest from a cell or tissue lysate or a body fluid for protein characterization or to investigate protein-protein interactions.
The process begins with an antibody, which has a high affinity and specificity for the target protein. This antibody is mixed with the sample, allowing antibody-target complexes to form. Any protein bound to the target protein also gets indirectly attached to the antibody in the process. Next, the solution is incubated with agarose beads, conjugated to a bacterial protein, which has a strong affinity for the constant region of antibodies. The bacterial protein binds to the antibody and connects the antibody- target complexes to the beads. Then, the solution is centrifuged to precipitate the beads, thereby extracting the entire complex containing the binding antibody, the target protein, and any interacting proteins. Finally, the bound proteins are extracted from the beads and released from each other and are used for further analysis by techniques such as Western blotting.
Several variations of different parts of this technique are commonly used, like pre-clearing, using peptide tags or magnetic beads, or analyzing other non-protein binding partners. IP can be preceeded by a pre-clearing step, to remove non-specific antibody-binding proteins in the sample and minimize background. This involves first incubating the sample with isotype control antibodies, allowing them to bind to these proteins, and then using agarose beads to precipitate the complexes. The sample is then ready to proceed to the actual IP.
Peptide tags are useful if a specific antibody is not available for IP. Here, the target protein can be genetically modified to contain a peptide epitope tag and an antibody against the tag is able to pull out the protein of interest. Magnetic beads are often used instead of agarose to precipitate the target. After binding to the antibody-target complex, the sample tube is placed in a strong magnetic field, which extracts the beads from the solution. This eliminates the need for centrifugation and improves speed and convenience.
Immunoprecipitation is also used for studying DNA or RNA binding proteins and are known as chromatin immunoprecipitation and RNA immunoprecipitation, respectively. These variations are useful for troubleshooting and adapting the method for different experimental applications. In this video, you will observe how to pre-clear a cell lysate and perform immunoprecipitation to extract a protein of interest, followed by Western blot analysis to validate the experiment.
To begin, place the pre-collected cells in a microcentrifuge and spin at 13 thousand rpm for three minutes. Following the spin, remove the supernatant and then resuspend the cells in 500 microliters of lysis buffer RIPA with PMSF. Now, disrupt the cells using a few quick pulses with a vortex and then aspirate the lysate a few times with a 25 gauge needle attached to a syringe, taking care to avoid creating bubbles. Place the cells on ice for 15 minutes. After incubating the samples on ice, centrifuge the lysate for 15 minutes at four degrees celsius.
Label a new 1.5 milliliter microcentrifuge tube. Following the spin, transfer the supernatant to the freshly labeled tube and discard the pellet. Next, pre-clear the lysate of contaminants that bind non-specifically to either the agarose beads or the primary antibody by adding 20 microliters of the Protein A/G PLUS-agarose beads and one microgram of an isotype control antibody to the lysate, which in this example is a mouse IgG1 isotype control. Incubate the tube on a rotator in a cold room for 30 minutes. After rotating the lysate in the cold room for 30 minutes, centrifuge the sample at 3200 rpm for 30 seconds at four degrees celsius. Remove the tube from the centrifuge and transfer the pre-cleared supernatant to a fresh labeled 1.5 milliliter microcentrifuge tube. Discard the pellet.
Now, determine the protein concentration of the cell lysate by performing a Bradford assay. Label seven 1. 5-milliliter microcentrifuge tubes one through six and sample and aliquot 1000 microliters of the Bradford reagent into each tube. Six of the tubes will be used to make a standard curve by adding various amounts of known quantities of BSA to each tube. The amounts to add are listed in this table. In the seventh sample tube, add one microliter of the pre-cleared lysate. Place 200 microliters from each of the seven tubes into individual wells of a flat-bottom 96-well plate, repeating each sample in triplicate so that there are three columns of seven samples. Read the plate on a plate reader, using a wavelength of 595 nanometers. After creating a standard curve in Excel, calculate the protein concentration of the pre-cleared lysate.
Next, label two 1.5-milliliter microcentrifuge tubes- one as control and the other as test, which in this example, will be the c-myc antibody. Place 500 micrograms of the pre-cleared lysate into each of these tubes and then bring the total volume for each tube up to 500 microliters using lysis buffer. Next, add two micrograms of the anti-c-myc antibody to the test group tube. For the control, add two micrograms of the mouse IgG1 isotype control antibody. Once the antibodies are added to the tubes, place the samples on a rotator in a cold room and incubate for two hours. Now, add the agarose beads. To do this, it is recommended to cut off the end of a pipette tip and then, using this modified tip, add 200 microliters of the Protein A/G PLUS-agarose beads to each tube. Incubate the tubes on a rotator in the cold room overnight.
Following the incubation, remove the tubes from the rotator and spin the lysates in the microcentrifuge to pull down the beads. After the spin is complete, remove the tubes from the centrifuge and aspirate the supernatant from each tube. Next, wash the beads using 500 microliters of 1X Dulbecco's PBS. Place the tubes in a microcentrifuge and spin down for 30 seconds at four degrees celsius. Following this, remove the supernatant. Repeat the wash and centrifuge steps one more time for a total of two times. Remove the tubes from the microcentrifuge and aspirate the buffer from each tube. Using gel loading tips, remove any left over buffer from the beads, keeping the beads on ice to elute the bound protein.
In this example, the protein is eluted into SDS-PAGE running buffer by boiling for Western blot analysis. To do this, resuspend the beads in 20 microliters of SDS-PAGE loading dye containing beta-mercaptoethanol, or BME. Boil the samples at 95 degrees celsius for five minutes to dissociate the immunocomplexes from the beads. Then, centrifuge the beads at maximum speed for 10 seconds at room temperature. Remove the tubes from the microcentrifuge and hold them in a rack at room temperature. Using gel loading tips, carefully pipette the samples from the beads and load them into wells of a 4 to 15% gradient SDS-PAGE gel. In addition to the samples, load a lane with a protein ladder as well as a lane with the pre-cleared lysate to serve as a loading control. Once the gel is loaded, run the gel at 100 volts.
After the dye front has reached the bottom of the gel, which should take approximately one hour, stop the gel and make a Western blot sandwich, ensuring that the PVDF membrane is between the gel and the cathode. Place the Western blot sandwich in the transfer apparatus and transfer the proteins on the gel to the membrane for one hour at 100 volts. After the transfer is complete, place the membrane in five milliliters of block to prevent the antibodies from binding non-specifically to the membrane. Rock at a low setting for an hour at room temperature. When the timer sounds, remove the blocking buffer. Add five milliliters of the blocking buffer with the detection antibody to the membrane. Here, an anti-c-myc antibody, that is different than the one used for the pull down, is used.
Incubate the blot over night, at four degrees celsius on a rocker at a low setting. Following the incubation, remove the antibody and blocking buffer. Wash the blot, using five milliliters of TBST for five minutes at room temperature, on a rocker at a low setting. This wash step should be repeated two to five times for a total of three to six washes, using fresh TBST for each wash. Add five milliliters of one to 1000 secondary antibody and blocking buffer to the blot. In this case, the secondary antibody is HRP-tagged anti-rabbit light chain. Incubate the blot on a rocker at a low setting for one our at room temperature. Next, remove the buffer and wash the blot with five milliliters of TBST. Incubate this wash on a rocker at a low setting for five minutes at room temperature. Repeat this wash for a total of six to 12 washes, each with a fresh five milliliters of TBST. Remove the final wash by first pouring the liquid off of the blot. Then, using tweezers, dab the edge of the blot on a laboratory wipe to remove any excess liquid and then place the blot in a fresh container. Next, cover the blot with 1X Chemiluminescent Detection Reagent and incubate for one minute.
Working quickly, dab the edge of the blot on a laboratory wipe to remove any excess detection reagent and then place the blot on the imaging surface of the Imager tray. Image using the Chemiluminescent program to capture multiple time points from 10 to 30 seconds. After the blot is imaged, choose an image with optimal band visibility and then export that image. Prior to moving the blot, use the Imager to take a picture of the blot to capture the location of the ladder. Then, export that image also. Finally, using a slide preparation software, such as PowerPoint, align the bands and ladder images to form one image.
This image shows the Western blot result for immunoprecipitation of the protein c-myc from thymocyte cells. From left to right, the lanes represent the isotype control, the c-myc IP, and the pre-cleared lysate input. The lane on the extreme right is a merged image of the molecular weight ladder. The strong band, at around 25 kilodaltons is from the light chain and the one at 50 kilodaltons is from the heavy chain of the binding antibody and are non-specific to the IP or the samples. C-myc runs around 67 kilodaltons on Western blots and is usually visible just below the 75 kilodalton ladder band. In this blot, the c-myc band is visible in the second lane but absent in the first lane, indicating that the IP antibody successfully pulled down c-myc. There is no visible band in the pre-cleared lysate lane, suggesting that this protein has low endogenous expression levels.
Subscription Required. Please recommend JoVE to your librarian.
Results
The results of the procedure detailed above are shown in Figure 2. From left to right, the lanes contain the control group (isotype), the test group (c-myc), the pre-cleared lysate (lysate), and the molecular weight ladder (ladder). The 25 and 75 kDa ladder bands are marked. The two prominent bands at ~25 kDa and 50 kDa are the light and heavy chain of the binding antibody, respectively and are non-specific to the IP or the samples. c-myc protein which runs around 67kDa on Western blots and is usually visible just below the 75 kDa ladder band. In this blot, the c-myc band is visible in the second lane, but absent in the first lane, indicating that the IP antibody successfully pulled down c-myc. There is no visible band in the pre-cleared lysate lane, suggesting that this protein has low endogenous expression levels.

Figure 2: Results of a Western Blot Analysis, used to assess the purification of c-myc by immunoprecipitation. A band at 67 kDa, corresponding to c-myc, is visible in the anti-c-myc lane, but not the isotype control lane. Note that c-myc levels were not high enough to be visualized in the lysate lane. Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
In short, immunoprecipitation is the isolation of a specific protein using an antibody. In this example, the results of the immunoprecipitation were analyzed by Western blot to assess the purity. The isolated protein could be used in a number of applications afterwards including: NMR for protein structure, Mass Spectrometry for amino acid sequence, or in vitro assays for enzymatic characterization. IPs can also characterize the interacting partners of proteins. For instance, following isolation, DNA or RNA could be isolated for sequencing. Co-immunoprecipitations assess protein-protein interactions. When the target protein is pulled down during an IP, interacting proteins can also be pulled down. These interacting partners can be assessed by mass spectrometry and Western blot. Immunoprecipitation is a powerful technique for studying protein biology.
Subscription Required. Please recommend JoVE to your librarian.
References
- Olliver, C. L. and Boyd, C. D. (1984). Immunoprecipitation of In Vitro Translation Products with Protein A Bound to Sepharose. In J. M. Walker (eds), Nucleic Acids. Methods in Molecular Biology (pp. 157-160). New Jersey: Humana Press.
- Thurston, C. F. and Henley, L. F. (1988). Direct Immunoprecipitation of Protein. In J. M. Walker (eds), New Protein Techniques. Methods in Molecular Biology (pp. 149-158). New Jersey: Humana Press.
- Anderson, N. G. (1998). Co-immunoprecipitation: Identification of Interacting Proteins. In R. A. Clegg (eds), Protein Targeting Protocols.Methods in Molecular Biology (pp. 35-45). New Jersey: Humana Press.
- Jackson, D. I. and Dickson, C. (1999). Protein Techniques: Immunoprecipitation, In Vitro Kinase Assays, and Western Blotting. In P.T. Sharpe and I. Mason (eds), Molecular Embryology. Methods in Molecular Biology (pp. 699-708). New Jersey: Humana Press.
- Trieu, E. P. and Targoff, I. N. (2015). Immunoprecipitation: Western Blot for Proteins of Low Abundance. In B. Kurien and R. Scofield (eds), Western Blotting. Methods in Molecular Biology (pp. 327-342). New York, NY: Humana Press.