

Summary
Il protocollo qui descritto fornisce istruzioni dettagliate su come analizzare le regioni genomiche di interesse per il potenziale di codifica delle microproteine utilizzando PhyloCSF sul browser del genoma UCSC di facile utilizzo. Inoltre, si raccomandano diversi strumenti e risorse per studiare ulteriormente le caratteristiche di sequenza delle microproteine identificate per ottenere informazioni sulle loro presunte funzioni.
Abstract
Il sequenziamento di nuova generazione (NGS) ha spinto in avanti il campo della genomica e ha prodotto sequenze di genoma intero per numerose specie animali e organismi modello. Tuttavia, nonostante questa ricchezza di informazioni sulla sequenza, gli sforzi completi di annotazione genica si sono dimostrati impegnativi, specialmente per le piccole proteine. In particolare, i metodi convenzionali di annotazione delle proteine sono stati progettati per escludere intenzionalmente le proteine putative codificate da brevi frame di lettura aperti (SORF) di lunghezza inferiore a 300 nucleotidi per filtrare il numero esponenzialmente più elevato di SORF non codificanti spuri in tutto il genoma. Di conseguenza, centinaia di piccole proteine funzionali chiamate microproteine (<100 amminoacidi di lunghezza) sono state erroneamente classificate come RNA non codificanti o completamente trascurate.
Qui forniamo un protocollo dettagliato per sfruttare strumenti bioinformatici gratuiti e pubblicamente disponibili per interrogare le regioni genomiche per il potenziale di codifica delle microproteine basato sulla conservazione evolutiva. In particolare, forniamo istruzioni dettagliate su come esaminare la conservazione della sequenza e il potenziale di codifica utilizzando le frequenze di sostituzione filogenetica dei codone (PhyloCSF) sul browser del genoma dell'Università della California Santa Cruz (UCSC). Inoltre, descriviamo in dettaglio i passaggi per generare in modo efficiente allineamenti di più specie di sequenze di microproteine identificate per visualizzare la conservazione della sequenza di aminoacidi e raccomandiamo risorse per analizzare le caratteristiche delle microproteine, comprese le strutture di dominio previste. Questi potenti strumenti possono essere utilizzati per aiutare a identificare presunte sequenze di codifica microproteica in regioni genomiche non canoniche o per escludere la presenza di una sequenza di codifica conservata con potenziale traslazionale in una trascrizione non codificante di interesse.
Introduction
L'identificazione del set completo di elementi codificanti nel genoma è stato un obiettivo importante sin dall'inizio del Progetto Genoma Umano, e rimane un obiettivo centrale verso la comprensione dei sistemi biologici e l'eziologia delle malattie a base genetica 1,2,3,4. I progressi nelle tecniche NGS hanno portato alla produzione di sequenze di genoma intero per un vasto numero di organismi, tra cui vertebrati, invertebrati, lieviti e piante5. Inoltre, i metodi di sequenziamento trascrizionale ad alto rendimento hanno ulteriormente rivelato la complessità del trascrittoma cellulare e identificato migliaia di nuove molecole di RNA con funzioni sia codificanti che non codificanti proteine 6,7. La decodifica di questa grande quantità di informazioni sulla sequenza è un processo continuo e le sfide rimangono con gli sforzi completi di annotazione genica8.
Il recente sviluppo di metodi di profilazione traslazionale, tra cui il profilo dei ribosomi 9,10 e il sequenziamento dei poli-ribosomi11, hanno fornito prove che indicano che centinaia di eventi di traduzione non canonica mappano a SORF attualmente non annotate in tutto il genoma, con il potenziale di generare piccole proteine chiamate microproteine o micropeptidi 12,13,14,15,16, 17. Le microproteine sono emerse come una nuova classe di proteine versatili precedentemente trascurate dai metodi standard di annotazione genica a causa delle loro piccole dimensioni (<100 amminoacidi) e della mancanza di caratteristiche genetiche classiche che codificano proteine 8,12,18,19,20. Le microproteine sono state descritte praticamente in tutti gli organismi, compresi i lieviti21,22, le mosche 17,23,24 e i mammiferi 25,26,27,28, e hanno dimostrato di svolgere ruoli critici in diversi processi, tra cui sviluppo, metabolismo e segnalazione dello stress 19,20,29, 30,31,32,33,34. Pertanto, è imperativo continuare a estrarre il genoma per ulteriori membri di questa classe a lungo trascurata di piccole proteine funzionali.
Nonostante il diffuso riconoscimento dell'importanza biologica delle microproteine, questa classe di geni rimane ampiamente sottorappresentata nelle annotazioni del genoma e la loro accurata identificazione continua ad essere una sfida continua che ha ostacolato i progressi nel campo. Recentemente sono stati sviluppati vari strumenti computazionali e metodi sperimentali per superare le difficoltà associate all'identificazione delle sequenze di codifica delle microproteine (ampiamente discusse in diverse revisioni complete 8,35,36,37). Molti recenti studi di identificazione delle microproteine 38,39,40,41,42,43,44,45,46,47 hanno fatto molto affidamento sull'uso di uno di questi algoritmi chiamato PhyloCSF 48,49 , un potente approccio di genomica comparativa che può essere sfruttato per distinguere le regioni del genoma che codificano proteine conservate da quelle che non sono codificanti.
PhyloCSF confronta le frequenze di sostituzione dei codon (CSF) utilizzando allineamenti nucleotidici multi-specie e modelli filogenetici per rilevare le firme evolutive dei geni che codificano per le proteine. Questo approccio empirico basato su modelli si basa sulla premessa che le proteine sono principalmente conservate a livello di amminoacidi piuttosto che alla sequenza nucleotidica. Pertanto, le sostituzioni di codone sinonimi, che codificano lo stesso amminoacido, o le sostituzioni di codone ad amminoacidi con proprietà conservate (cioè carica, idrofobicità, polarità) sono valutate positivamente, mentre le sostituzioni non sinonimi, comprese le sostituzioni missense e senza senso, ottengono un punteggio negativo. PhyloCSF è addestrato su dati dell'intero genoma e ha dimostrato di essere efficace nel segnare brevi porzioni di una sequenza codificante (CDS) in isolamento dalla sequenza completa, che è necessaria quando si analizzano microproteine o singoli esoni di geni codificanti proteine standard48,49.
In particolare, la recente integrazione degli hub di traccia PhyloCSF nel Genome Browser 49,50,51 dell'Università della California Santa Cruz (UCSC) consente ai ricercatori di ogni provenienza di accedere facilmente a un'interfaccia user-friendly per interrogare le regioni genomiche di interesse per il potenziale di codifica delle proteine. Il protocollo descritto di seguito fornisce istruzioni dettagliate su come caricare gli hub di traccia PhyloCSF sul browser del genoma UCSC e successivamente interrogare le regioni genomiche di interesse per sondare le regioni codificanti proteine ad alta confidenza (o la loro mancanza). Inoltre, nel caso in cui si osservi un punteggio PhyloCSF positivo, vengono delineati passaggi per analizzare ulteriormente il potenziale di codifica delle microproteine e generare in modo efficiente allineamenti di più specie delle sequenze di amminoacidi identificate per illustrare la conservazione delle sequenze tra specie. Infine, diverse risorse e strumenti aggiuntivi disponibili pubblicamente sono introdotti nella discussione per esaminare le caratteristiche delle microproteine identificate, comprese le strutture di dominio previste e le informazioni sulla presunta funzione delle microproteine.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Il protocollo descritto di seguito descrive i passaggi per caricare e navigare le tracce del browser PhyloCSF sul browser del genoma UCSC (generato da Mudge et al.49). Per domande generali riguardanti il browser del genoma UCSC, una guida per l'utente del browser Genome completa può essere trovata qui: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. Caricamento dell'hub di traccia PhyloCSF sul browser del genoma UCSC
- Aprire una finestra del browser Internet e passare al browser del genoma UCSC (https://genome.ucsc.edu/).
- Sotto l'intestazione I nostri strumenti , seleziona l'opzione Traccia hub .
NOTA: l'opzione Track Hubs si trova anche nella scheda I miei dati . - Nella scheda Hub pubblici digitare PhyloCSF nella casella Termini di ricerca . Fare clic sul pulsante Cerca hub pubblici .
- Connettiti a PhyloCSF facendo clic sul pulsante Connetti per il PhyloCSF del nome hub (Descrizione: Potenziale evolutivo di codifica delle proteine misurato da PhyloCSF).
NOTA: questo Track Hub verrà caricato su numerosi assiemi, tra cui umano (hg19 e hg38) e mouse (mm10 e mm39). - Dopo aver fatto clic su Connetti, attendere di essere reindirizzati alla pagina UCSC Genome Browser Gateway (https://genome.ucsc.edu/cgi-bin/hgGateway).
2. Navigazione verso i geni di interesse utilizzando gli identificatori genici
- Selezionare l'assembly di specie e genoma da interrogare. Per interrogare una specie diversa (ad esempio, il mouse), selezionare la specie di interesse sotto l'intestazione Sfoglia/Seleziona specie facendo clic sull'icona appropriata o digitare la specie nella casella di testo che dice Inserisci specie, nome comune o ID assemblea.
NOTA: l'assieme è elencato direttamente sotto l'intestazione Trova posizione (Find Position ). In genere, l'impostazione predefinita è l'Assembly umano (ad esempio, dicembre 2009 [GRCh37/hg19]). - Scegliete l'assieme da cercare sotto l'intestazione Trova posizione (Find Position ) utilizzando il menu a discesa.
- Inserisci la posizione, il simbolo del gene o i termini di ricerca nella casella Posizione/Termine di ricerca e fai clic su Vai per accedere a un gene di interesse sul Browser del genoma.
- Se la ricerca ha portato a più corrispondenze, attendi di essere reindirizzato a una pagina che richiede la selezione di una posizione di interesse. Fare clic sul gene di interesse appropriato.
3. Navigazione verso le regioni genomiche di interesse utilizzando le informazioni di sequenza
- Passare al browser del genoma UCSC (https://genome.ucsc.edu/) e selezionare lo strumento di allineamento simile a BLAST (BLAT) sotto l'intestazione I nostri strumenti per interrogare una specifica sequenza di DNA o proteine. In alternativa, passa il cursore sulla scheda Strumenti e seleziona l'opzione Blat o segui questo link: https://genome.ucsc.edu/cgi-bin/hgBlat.
- Seleziona la specie (Genoma) e l'Assemblaggio di interesse utilizzando i menu a discesa.
- Definire il tipo di query utilizzando il menu a discesa.
- Incollare la sequenza di interesse nella casella di testo BLAT Search Genome e fare clic su Invia.
- Fare clic sul collegamento del browser sotto l'intestazione AZIONI per navigare verso la regione genomica di interesse.
4. Identificazione delle sORF conservate utilizzando i dati di traccia PhyloCSF
- Scansiona visivamente l'area genomica di interesse per ottenere un punteggio positivo nelle regioni PhyloCSF (Figura 1).
NOTA: per una spiegazione dettagliata di come interpretare visivamente i punteggi PhyloCSF sul browser del genoma UCSC, vedere la sezione dei risultati rappresentativi di seguito. - Utilizzate la funzione di zoom per ingrandire le regioni di interesse per esaminare le caratteristiche della sequenza e cercare codoni start/stop. Per ingrandire manualmente, tieni premuto il tasto Maiusc e fai clic e tieni premuto il pulsante del mouse mentre trascini lungo l'area di interesse. In alternativa, utilizzare i pulsanti di zoom avanti e indietro nella parte superiore della pagina per navigare (sono disponibili opzioni di zoom 1,5x, 3x, 10x o di base).
NOTA: Prima di utilizzare i pulsanti di zoom in/zoom out , è necessario riposizionare il gene in modo che la regione di interesse si trovi al centro dello schermo. Per eseguire questa azione, fare clic sull'immagine e trascinarla a sinistra o a destra per spostare la regione genomica orizzontalmente come desiderato o utilizzare le frecce di spostamento nella parte superiore della pagina. - Ingrandire fino a quando la sequenza nucleotidica (base) è visibile.
NOTA: La sequenza nucleotidica apparirà direttamente sopra il punteggio +1 Smoothed PhyloCSF. - Scansiona visivamente la sequenza nucleotidica vicino all'inizio e alla fine delle regioni PhyloCSF con punteggio positivo per identificare i codoni putativi start (ATG) e stop (TGA / TAA / TAG).
NOTA: Se il gene di interesse si trova sul filamento meno del DNA, i codoni di inizio e di arresto saranno il complemento inverso (cioè CAT per il codone di partenza e TCA / TTA / CTA per il codone di arresto).
5. Visualizzazione di regioni omologhe in altri genomi
- Passa il mouse sopra l'intestazione Visualizza nella parte superiore della pagina e fai clic sull'opzione In altri genomi (Converti ).
- Definisci il genoma di interesse utilizzando il menu a discesa sotto l'intestazione Nuovo genoma .
- Selezionare l'assemblaggio genomico di interesse utilizzando il menu a discesa sotto l'intestazione Nuovo assemblaggio , quindi fare clic sul pulsante Invia .
- Una volta che il browser restituisce un elenco di regioni nel nuovo assieme con somiglianza, fare clic sul collegamento posizione cromosomica per passare alla regione omologica di interesse.
NOTA: la percentuale di basi totali (nucleotidi) e l'intervallo coperto dalla regione saranno definiti per ogni regione elencata. Maggiore è la percentuale di basi corrispondenti, maggiore è la conservazione per la regione di interesse. - Seguire le stesse strategie di navigazione descritte nella Sezione 4 per analizzare la sequenza.
6. Generazione di allineamenti di sequenze multi-specie per microproteine di interesse
- Fare clic sul gene di interesse nella traccia GENCODE sul browser del genoma UCSC (indicato nella Figura 1A con una casella blu) per accedere alla pagina di descrizione del gene.
- Sotto l'intestazione Sequenza e collegamenti a strumenti e database , fare clic sul collegamento nella tabella che legge Altre specie FASTA.
- Clicca sulle caselle associate alle specie di interesse per selezionarle. Clicca su Invia. Copia e incolla le sequenze visualizzate nella parte inferiore della pagina in formato FASTA in un documento di elaborazione testi.
- Aprire una seconda finestra del browser e passare allo strumento Clustal Omega Multiple Sequence Alignment 52 sul sito web dell'Istituto europeo di bioinformatica (EMBL-EBI)53,54: https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Incollare i file di sequenza che si trovano ancora negli Appunti nella casella in STEP 1 che legge le sequenze in qualsiasi formato supportato. Scorri fino in fondo alla pagina e fai clic su Invia. Guarda sotto i risultati allineati (in carattere nero) per i simboli che indicano il grado di conservazione di ciascun amminoacido (i simboli sono definiti nella Tabella 1).
NOTA: la generazione dell'allineamento potrebbe richiedere alcuni minuti. - Per visualizzare le proprietà degli amminoacidi a colori, fare clic sul collegamento Mostra colori direttamente sopra le sequenze per colorare gli amminoacidi in base alle loro proprietà (definite nella Tabella 2).
- Copiare e incollare l'allineamento della sequenza in un programma di elaborazione testi o presentazione per generare un file di figura o illustrazione (ad esempio, Figura 2).
NOTA: utilizzare un carattere monospaziato per l'allineamento, ad esempio Courier. - Per visualizzare altri output dalla pagina dei risultati di Clustal Omega , fare clic sulle schede appropriate (ad esempio, Albero guida o Albero filogenetico).
- Fare clic sulla scheda Visualizzatori risultati per visualizzare le informazioni sulla sequenza utilizzando Jalview, un programma gratuito specializzato in modifica, visualizzazione e analisi dell'allineamento di sequenze multiple55, o per accedere a collegamenti diretti a MView e Simple Phylogeny56.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Qui useremo la microproteina convalidata mitoregulina (Mtln) come esempio per dimostrare come un sORF conservato genererà un punteggio PhyloCSF positivo che può essere facilmente visualizzato e analizzato sul browser del genoma UCSC. La mitoregulina è stata precedentemente annotata come RNA non codificante (precedentemente gene umano ID LINC00116 e gene di topo ID 1500011K16Rik). La genomica comparativa e i metodi di analisi della conservazione delle sequenze hanno svolto un ruolo fondamentale nella sua scoperta iniziale 40,57,58,59,60,61, evidenziando la forza di questi metodi. Per questo esempio, verrà utilizzato l'assembly GRCm38/mm10 (dicembre 2011) del mouse. La ricerca può essere eseguita utilizzando gli identificatori genetici (mitoregulina, Mtln) o la posizione del gene (chr2:127.791.364-127.792.496) come descritto nella sezione 2 del protocollo. In alternativa, la sequenza aminoacidica per mitoregulina (mostrata nella Figura 2) può essere ricercata utilizzando lo strumento BLAT (descritto nella sezione 3 del protocollo).
Apparirà una schermata simile a quella raffigurata nella Figura 1A con il PhyloCSF Track Hub visibile nella parte superiore dello schermo. Le tracce Smoothed PhyloCSF (levigate con un modello di Markov nascosto che definisce una probabilità che ogni codone stia codificando) sono rappresentate come sei tracce totali, con tre tracce corrispondenti al filamento più di DNA (raffigurato in verde come PhyloCSF +1, +2 e +3) e tre tracce corrispondenti al filamento meno di DNA (raffigurato in rosso come PhyloCSF -1, -2 e -3). Queste tracce rappresentano i tre potenziali frame di lettura per il gene di interesse in ciascuna direzione. Nella finestra del browser, gli esoni sono raffigurati come rettangoli blu collegati da sottili linee orizzontali blu, che rappresentano gli introni. Le punte di freccia sulle regioni introniche indicano in quale direzione viene trascritto il gene (e quindi, su quale filamento concentrarsi per il punteggio PhyloCSF). Per l'esempio di Mtln nella Figura 1, le punte di freccia introniche puntano a sinistra. Pertanto, il gene Mtln viene trascritto dal filamento meno del DNA e il punteggio PhyloCSF pertinente è rappresentato nelle tracce -1, -2 e -3 (in rosso).
Ogni traccia PhyloCSF è raffigurata come una sottile linea nera con regioni di punteggio negativo raffigurate in verde chiaro / rosso sotto la linea e regioni di punteggio positivo indicate in verde scuro / rosso sopra la linea. Come descritto nell'introduzione, un punteggio PhyloCSF positivo indica una regione conservata che è probabilmente codificata. Si noti che per le regioni codificanti proteine con una conservazione della sequenza particolarmente elevata, spesso ottengono anche un punteggio positivo sul filamento antisenso; tuttavia, il punteggio PhyloCSF è solitamente più alto sul filo corretto. Ad esempio, questo può essere visto nella Figura 1 per Mtln dove la sequenza di codifica corretta ha punteggi molto alti nella traccia PhyloCSF -1 e anche il filamento antisenso (traccia PhyloCSF +2) genera un punteggio positivo. Come si vede nella Figura 1A (indicata con la scatola nera), c'è una regione nel primo esone di Mtln che ha un punteggio molto alto sulla traccia PhyloCSF -1, suggerendo che questo potrebbe corrispondere a una regione di codifica. Per esaminare questa regione in modo più dettagliato, è utile ingrandire e ingrandire la regione (Figura 1B). Come mostrato nella Figura 1C,D, la regione di punteggio positivo nel primo esone di Mtln inizia direttamente su un codone di partenza (Figura 1C) e termina in un codone di arresto (Figura 1D), il che indica che questo ORF è altamente conservato e suggerisce fortemente che si tratta di un ORF codificante. Poiché Mtln si trova sul filamento meno del DNA, i codoni di inizio e di arresto sono mostrati come il complemento inverso del codone (cioè, il codone di inizio ATG è mostrato come CAT [Figura 1C] e il codone di arresto TGA è mostrato come TCA [Figura 1D]).
Oltre a utilizzare PhyloCSF per cercare regioni conservate con potenziale di codifica microproteica, questa tecnica può anche essere applicata come analisi di primo passaggio di presunti RNA non codificanti per escludere la presenza di un ORF conservato, fornendo così supporto per un'annotazione non codificante. Ad esempio, l'analisi del lncRNA HOTAIR62,63 ben caratterizzato utilizzando PhyloCSF mostra un punteggio negativo in tutto il gene in tutte e sei le tracce (Figura 3), indicando fortemente una mancanza di conservazione della sequenza e fornendo supporto che HOTAIR è correttamente annotato come RNA non codificante.
Come chiaramente visto nella Figura 1, l'intero ORF codificante per mitoregulina si trova all'interno di un singolo esone, producendo così una lettura semplice e diretta da parte di PhyloCSF con una singola regione ininterrotta e con punteggio positivo. Tuttavia, i dati dell'hub di traccia PhyloCSF non sono sempre così chiari e facili da interpretare. Ad esempio, la microproteina mitolamban/Stmp1/Mm47 codificata dal gene 1810058I24Rik del topo 47,64,65 raffigura un ORF conservato che si estende su tre esoni (Figura 4A), e il punteggio PhyloCSF positivo salta dalla traccia +2 nell'esone 1 (Figura 4B) alla traccia +3 nell'esone 2 (Figura 4C), e poi di nuovo alla traccia +2 nell'esone 3 (Figura 4D ). Mentre a prima vista questo sembra confuso, la spiegazione è abbastanza semplice. PhyloCSF segna i sei potenziali frame di lettura (tre sul filamento più di DNA e tre sul filamento meno) delle regioni genomiche senza considerare l'architettura specifica dell'esone / introne per ciascun gene. Pertanto, conserva le informazioni sulla sequenza intronica nella periodicità a 3 nucleotidi dei fotogrammi di lettura. Quindi, se un introne contiene un numero di nucleotidi che non è divisibile per tre (cioè tre nucleotidi / codone), il frame di lettura PhyloCSF salterà da una traccia all'altra.
Infine, PhyloCSF può anche essere efficacemente utilizzato per identificare più ORF codificanti distinti all'interno di una singola molecola di RNA. Ad esempio, la microproteina MIEF1 (MIEF1-MP) è codificata all'interno dell'UTR 5' del fattore di allungamento mitocondriale 1 (MIEF1)66 (Figura 5). Quando la regione genomica MIEF1 viene analizzata da PhyloCSF, un punteggio PhyloCSF discreto positivo corrispondente al MIEF1-MP (Figura 5C) può essere facilmente osservato a monte del CDS principale per MIEF1 (Figura 5B). Un'ulteriore discussione sul MIEF1 e sulla sua microproteina associata (MIEF1-MP) è fornita di seguito nella discussione insieme a un riepilogo dei punti di forza e di debolezza dei metodi e dei protocolli delineati in questo articolo.

Figura 1: L'analisi PhyloCSF del gene mitoregulina (Mtln) indica una regione di conservazione ad alta sequenza corrispondente a una microproteina convalidata. (A) Gli screenshot del browser del genoma UCSC e delle tracce PhyloCSF mostrano che Mtln contiene due esoni e un singolo introne. Le punte di freccia all'interno dell'introne puntano a sinistra, indicando che il gene Mtln viene trascritto dal filamento meno del DNA, e i relativi punteggi PhyloCSF sono quindi mostrati nelle tracce -1, -2 e -3 (in rosso). La sequenza completa di codifica della mitoregulina è contenuta all'interno dell'esone 1 e ha un punteggio elevato nella traccia PhyloCSF -1 (B). Un codone di partenza conservato può essere chiaramente osservato all'inizio della regione con punteggio positivo nella traccia PhyloCSF -1 (C), che è evidenziata con una casella verde (CAT, complemento inverso ATG). Inoltre, un codone di arresto conservato (TCA, complemento inverso TGA) è indicato con una casella rossa nel pannello (D), che si allinea con la fine della regione PhyloCSF con punteggio positivo. Informazioni dettagliate sul gene Mtln possono essere trovate cliccando sull'identificatore del gene Mtln all'interno della casella blu (mostrata nel pannello A). Da notare, le regioni codificanti proteine altamente conservate spesso ottengono anche un punteggio positivo sul filamento antisenso (visto qui nella traccia PhyloCSF +2 per Mtln). Tuttavia, il punteggio PhyloCSF è in genere più alto sul filamento corretto (la traccia PhyloCSF -1 in questo esempio). Fare clic qui per visualizzare una versione più grande di questa figura.

Figura 2: Allineamento di sequenze di specie multiple della microproteina mitoregulina generata utilizzando il programma Clustal Omega. Le sequenze di aminoacidi mitoreguline per le otto specie indicate sono state estratte come dettagliato nella sezione 6 del protocollo e allineate con lo strumento di allineamento a sequenza multipla Clustal Omega. Le proprietà degli amminoacidi sono indicate dal colore (rosso, piccolo/idrofobo; blu, acido; magenta, basico; verde, idroxl/sulfidril/ammina) (ulteriormente definito nella Tabella 2). I simboli sotto gli amminoacidi indicano il grado di conservazione (asterischi, residui completamente conservati; due punti, amminoacidi con proprietà fortemente simili; periodi, conservazione tra gruppi di proprietà debolmente simili) (ampiamente dettagliato nella Tabella 1). Fare clic qui per visualizzare una versione più grande di questa figura.
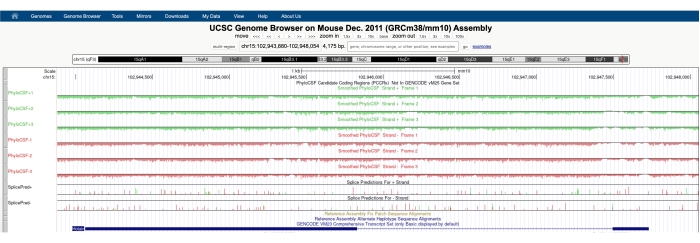
Figura 3: Uno screenshot delle tracce PhyloCSF per l'RNA non codificante lungo convalidato Hotair mostra una mancanza di conservazione della sequenza in tutto il suo locus genomico. Le punte di freccia nella regione intronica di Hotair puntano a sinistra, indicando che l'lncRNA è trascritto dal filamento negativo del DNA, e quindi le tracce PhyloCSF -1, -2 e -3 dovrebbero essere al centro dell'analisi. Si noti che il punteggio PhyloCSF è negativo in tutto il gene (per tutte e sei le tracce), indicando una mancanza di conservazione della sequenza, che supporta la sua corretta annotazione come RNA non codificante. Fare clic qui per visualizzare una versione più grande di questa figura.

Figura 4: Analisi PhyloCSF del gene 1810058I24Rik del topo, che codifica per la microproteina mitolamban/Stmp1/Mm47. (A) Il gene 1810058I24Rik del topo è composto da tre esoni e le punte di freccia nelle regioni introniche puntano a destra, indicando che è trascritto sul filamento più del DNA e quindi le tracce PhyloCSF +1, +2 e +3 dovrebbero essere analizzate. La sequenza di codifica delle microproteine conservata copre tutti e tre gli esoni, iniziando nell'esone 1 (B), leggendo attraverso l'esone 2 (C) e terminando nell'esone 3 (D). Si noti che il punteggio PhyloCSF positivo si trova sulla traccia +2 nell'esone 1, sulla traccia +3 nell'esone 2 e sulla traccia +2 nell'esone 1. La ragione del movimento del punteggio positivo da una traccia all'altra è che PhyloCSF analizza i sei potenziali fotogrammi di lettura della sequenza di DNA indipendentemente dalla struttura esone/introne del gene. Pertanto, un introne contenente un numero di nucleotidi che non è divisibile per tre (tre nucleotidi / codone) causerà uno spostamento nel fotogramma di lettura su una traccia diversa. Fare clic qui per visualizzare una versione più grande di questa figura.

Figura 5: L'analisi del locus genomico Mief1 con PhyloCSF identifica una regione con potenziale di codifica proteica nell'UTR 5' che è indipendente dal principale CDS Mief1 sull'RNA condiviso. Questo ORF a monte conservato (uORF) ha dimostrato di codificare una microproteina chiamata Mief1-MP. (A) Panoramica del locus genomico Mief1 . Le punte di freccia negli introni puntano a destra, indicando che Mief1 è trascritto dal filamento più di DNA (focus sulle tracce PhyloCSF +1, +2 e +3 per determinare il potenziale di codifica). Il principale CDS Mief1 codifica per una proteina di 463 aminoacidi ed è mostrato nel pannello (B). Tuttavia, esiste anche un ORF a monte conservato distinto all'interno dell'UTR 5' di Mief1 che codifica per una microproteina unica di 70 amminoacidi chiamata Mief1-MP (C). Come visto nel Pannello C, il Mief1-MP ha il proprio codone di avvio e arresto conservato all'interno del Mief1 5' UTR, e l'ORF ha un punteggio molto alto sulla traccia PhyloCSF +1, fornendo una forte evidenza che codifica una microproteina funzionale. Abbreviazioni: ORF = cornice di lettura aperta; uORF = ORF a monte; UTR = regione non tradotta; CDS = sequenza di codifica. Fare clic qui per visualizzare una versione più grande di questa figura.
| Simbolo | Livello di conservazione degli aminoacidi | Aminoacidi raggruppati |
| Asterisco (*) | Residuo completamente conservato | Non applicabile (singolo residuo completamente conservato) |
| Colon (:) | Gruppi con proprietà fortemente simili | STA; NEQK; NHQK; NDEQ; QHRK; MILV; MILF; HY; FyW |
| Periodo (.) | Gruppi con proprietà debolmente simili | CSA; ATV; SAG; STNK; STPA; SGND; SNDEQK; NDEQHK; NEQHRK; FVLIM; HFY · |
| Spazio (nessun simbolo) | Nessuna somiglianza | Non applicabile (nessuna somiglianza) |
Tabella 1: Definizioni dei simboli di consenso per gli allineamenti a sequenza multipla generati da Clustal Omega. L'allineamento di sequenze di specie multiple mostrato nella Figura 2 è stato generato utilizzando Clustal Omega52. Abbreviazioni: serina (S), treonina (T), alanina (A), asparagina (N), acido glutammico (E), glutammina (Q), lisina (K), acido aspartico (D), arginina (R), metionina (M), isoleucina (I), leucina (L), fenilalanina (F), istidina (H), tirosina (Y), triptofano (W), cisteina (C), valina (V), glicina (G), prolina (P).
| Colore carattere | Proprietà | Residuo di aminoacidi [Abbreviazione] |
| Rosso | Piccolo, idrofobo | alanina [A], valina [V], fenilalanina [F], prolina [P], metionina [M], isoleucina [I], leucina [L], triptofano [W] |
| Blu | Acido | acido aspartico [D], acido glutammico [E] |
| Magenta | Basico | arginina [R], lisina [K] |
| Verde | Hydroxl, sulfidrile, ammina, +G | serina [S], treonina [T], tirosina [Y], istidina [H], cisteina [C], asparagina [N], glicina [G], glutammina [Q] |
Tabella 2: Proprietà degli amminoacidi raffigurati nella Figura 2. Clustal Omega52 è stato utilizzato per generare l'allineamento a sequenza multipla mostrato nella Figura 2.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Il protocollo qui presentato fornisce istruzioni dettagliate su come interrogare le regioni genomiche di interesse per il potenziale di codifica delle microproteine utilizzando PhyloCSF sul browser del genoma UCSC 48,49,50,51. Come descritto sopra, PhyloCSF è un potente algoritmo di genomica comparativa che integra modelli filogenetici e frequenze di sostituzione dei codone per identificare le firme evolutive tipiche dei geni codificanti proteine48,49. PhyloCSF è stato ampiamente utilizzato per identificare microproteine funzionali in regioni genomiche precedentemente annotate come non codificanti 38,39,40,41,42,43,44,45,46,47 , e questo approccio ha dimostrato di superare altri metodi di genomica comparativa per brevi sequenze come microproteine piccole come 13 amminoacidi e per piccoli esoni di proteine canoniche 35,48,49. In particolare, l'utilità di PhyloCSF come metodo robusto per identificare sequenze funzionali di codifica proteica attraverso la conservazione evolutiva si estende oltre quella delle specie di vertebrati e invertebrati ed è stata persino recentemente applicata ai genomi virali per interrogare con successo la capacità di codifica proteica del genoma SARS-CoV-267.
Oltre a identificare presunte sequenze codificanti all'interno di RNA non codificati annotati, un vantaggio di PhyloCSF è che può anche rilevare in modo affidabile microproteine conservate codificate da ORF all'interno di regioni annotate non tradotte (UTR) di geni codificanti proteine canoniche, inclusi ORF a monte e 3' a valle (ulRIF e dORF, rispettivamente)8,19,66,68 . Ad esempio, la microproteina MIEF1 (MIEF1-MP) è codificata nel 5' UTR del fattore di allungamento mitocondriale 1 (MIEF1)66. Nel caso di MIEF1-MP, un punteggio PhyloCSF discreto positivo corrispondente al MIEF1-MP è osservato a monte dell'ORF che codifica MIEF1 (Figura 5). Mentre alcune microproteine codificate uORF interagiscono direttamente con le proteine canoniche a valle sul loro mRNA condiviso (es. MIEF1-MP e MIEF1), altre funzionano indipendentemente dalla proteina codificata dal CDSprincipale 66,68. Pertanto, quando si caratterizzano microproteine codificate uORF, non si deve presumere che funzionino tramite la regolazione diretta del loro prodotto proteico a valle.
Mentre PhyloCSF ha molti chiari punti di forza come strumento per l'identificazione di sequenze di codifica microproteina conservate, è importante riconoscere diversi limiti di questo metodo. In primo luogo, mentre la conservazione della sequenza suggerisce fortemente che una regione genomica ha subito una selezione funzionale e quindi sta codificando, la mancanza di una conservazione robusta e un conseguente punteggio PhyloCSF negativo non esclude definitivamente il potenziale di codifica per una data sequenza. In altre parole, affidarsi esclusivamente a PhyloCSF può comportare la supervisione di ORF tradotti che non sono fortemente conservati ma producono comunque microproteine funzionali. In particolare, le regioni genomiche con bassi punteggi di conservazione o negativi potrebbero corrispondere a regioni codificanti specie-specifiche o a quelle di geni evolutivi "giovani" attraverso la divergenza di sequenza o la nascita di geni de novo 46,69,70,71,72,73,74. Ad esempio, la microproteina ASAP, che è codificata da quello che in precedenza si pensava fosse l'RNA umano non codificante LINC00467, non è valutata positivamente da PhyloCSF perché la sequenza di amminoacidi è conservata solo nei mammiferi superiori75. Inoltre, studi recenti hanno identificato diverse microproteine specifiche per l'uomo, tra cui una codificata dall'lncRNA intergenico RP3-527G5.1, che non genera un punteggio PhyloCSF positivo 68,72. A questo proposito, l'assenza di un punteggio PhyloCSF positivo non può essere interpretata come prova di una regione non codificante e deve essere interpretata con cautela.
Una seconda considerazione da tenere a mente quando si utilizza PhyloCSF è che anche se un punteggio positivo è altamente suggestivo di selezione funzionale e capacità di codifica proteica, questa linea di prove non può stare da sola e deve essere convalidata sperimentalmente. Esempi di metodi che possono essere utilizzati per generare prove a sostegno dell'espressione stabile di microproteine includono il rilevamento della proteina putativa mediante spettrometria di massa o western blotting utilizzando un anticorpo sollevato contro la sequenza di microproteine di interesse. In alternativa, poiché può essere difficile generare anticorpi affidabili per le microproteine a causa della mancanza di scelte di sequenza per un'antigenicità ottimale, è anche possibile utilizzare CRISPR/Cas9 e il percorso di riparazione diretta dall'omologia (HDR) per introdurre un tag epitopo nel locus endogeno nel frame con la presunta sequenza di microproteine, facilitando così il rilevamento della proteina di interesse utilizzando un anticorpo ad alta affinità (ad esempio, BANDIERA, HA, V5, Myc)18. Un'ultima limitazione di PhyloCSF da riconoscere è che, sebbene sia attualmente integrato in molti degli assemblaggi genomici comunemente usati, tra cui Homo sapiens (umano hg19, hg38), Mus musculus (topo mm10, mm39), Gallus gallus (pollo, galGal4, galGal6), Drosophila melanogaster (moscerino della frutta, dm6), Caenorhabditis elegans (nematodi, ce11) e SARS-CoV-2 (wuhCor1), ci sono ancora molte specie che attualmente non possono essere interrogate direttamente sul browser del genoma UCSC.
L'identificazione di domini conservati o caratteristiche di sequenza all'interno di microproteine identificate può aiutare ad aumentare la fiducia nella loro rilevanza funzionale e fornire alcune informazioni sulla loro funzione putativa. Qui forniamo raccomandazioni per strumenti e risorse specifici che possono essere utilizzati per analizzare le sequenze di aminoacidi microproteici identificati in modo più dettagliato per ottenere tali informazioni. Gli strumenti specifici elencati di seguito (e riassunti nella Tabella dei materiali) sono liberamente disponibili al pubblico e li abbiamo trovati particolarmente user-friendly e robusti negli studi sulle microproteine 18,38,39,40,41,47. Oltre agli strumenti qui descritti, ci sono una moltitudine di risorse aggiuntive che possono essere trovate nei portali di risorse bioinformatiche come Expasy (https://www.expasy.org) e EMBL-EBI (https://www.ebi.ac.uk/services/all). Tuttavia, la definizione dettagliata delle specifiche per ciascuno degli strumenti all'interno di questi repository esula dall'ambito di questo articolo. Qui raccomandiamo le seguenti risorse.
In primo luogo, TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) analizza le sequenze proteiche di interesse per la presenza di domini transmembrana. In particolare, un certo numero di microproteine che sono state funzionalmente caratterizzate finora contengono domini transmembrana a passaggio singolo, che facilitano la loro localizzazione nelle regioni di membrana e consentono la loro regolazione diretta di canali ionici, scambiatori ed enzimi associati alla membrana30. In secondo luogo, il National Center for Biotechnology Information (NCBI) Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) è uno strumento popolare utilizzato per identificare domini conservati all'interno di sequenze proteiche o nucleotidiche codificanti. In terzo luogo, il database della famiglia proteica (Pfam)78 (http://pfam.xfam.org) fornisce allineamenti e classificazioni di famiglie e domini proteici. In quarto luogo, WoLF PSORT79 (https://wolfpsort.hgc.jp/) è uno strumento che può essere impiegato per prevedere la localizzazione delle proteine subcellulari. Quinto, COXPRESdB80 è un database di co-espressione genica (https://coxpresdb.jp) che fornisce relazioni geniche co-regolate per stimare le funzioni geniche. Infine, SignalP 6.081 è un programma di previsione ampiamente utilizzato (https://services.healthtech.dtu.dk/service.php?SignalP) che riconosce la presenza di una sequenza peptidica di segnale e predice la posizione del sito di scissione.
In sintesi, i metodi qui descritti possono essere utilizzati per analizzare efficacemente le regioni genomiche di interesse per il potenziale di codifica delle proteine utilizzando PhyloCSF sul browser del genoma UCSC. Questi metodi sono altamente accessibili e possono essere facilmente appresi e applicati in modo efficiente da individui senza una precedente formazione o esperienza in bioinformatica o genomica comparativa. Come dimostrato qui in dettaglio, PhyloCSF è un potente strumento che può essere applicato come analisi di primo passaggio per aiutare a distinguere i geni che codificano le proteine rispetto ai geni non codificanti nei genomi di vertebrati, invertebrati e virali, e i punti di forza di questo approccio superano di gran lunga le debolezze note.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori dichiarano di non avere interessi finanziari concorrenti.
Acknowledgments
Questo lavoro è stato sostenuto da sovvenzioni del National Institutes of Health (HL-141630 e HL-160569) e della Cincinnati Children's Research Foundation (Trustee Award).
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
