

Summary
ここで説明するプロトコルは、ユーザーフレンドリーなUCSCゲノムブラウザ上のPhyloCSFを使用して、マイクロタンパク質コードの可能性について関心のあるゲノム領域を分析する方法に関する詳細な手順を提供します。さらに、同定されたマイクロタンパク質の配列特性をさらに調査し、推定機能に関する洞察を得るために、いくつかのツールとリソースが推奨されます。
Abstract
次世代シーケンシング(NGS)は、ゲノミクスの分野を前進させ、多数の動物種およびモデル生物の全ゲノム配列を生み出しました。しかし、この豊富な配列情報にもかかわらず、包括的な遺伝子アノテーションの取り組みは、特に小さなタンパク質では困難であることが証明されています。特に、従来のタンパク質アノテーション法は、ゲノム全体にわたって指数関数的に多い数の偽の非コードsORFを除外するために、長さが300ヌクレオチド未満の短いオープンリーディングフレーム(sORF)によってコードされる推定タンパク質を意図的に除外するように設計されていた。その結果、マイクロタンパク質(<100アミノ酸長)と呼ばれる何百もの機能的な小さなタンパク質が、誤ってノンコーディングRNAとして分類されたり、完全に見過ごされてきました。
ここでは、無料で公開されているバイオインフォマティクスツールを活用して、進化的保存に基づいてゲノム領域にマイクロタンパク質コードの可能性を照会するための詳細なプロトコルを提供します。具体的には、ユーザーフレンドリーなカリフォルニア大学サンタクルーズ校(UCSC)ゲノムブラウザの系統コドン置換頻度(PhyloCSF)を使用して配列保存とコーディングの可能性を調べる方法について、段階的な手順を提供します。さらに、同定されたマイクロタンパク質配列の複数種のアラインメントを効率的に生成してアミノ酸配列の保存を視覚化する手順を詳述し、予測されたドメイン構造を含むマイクロタンパク質特性を分析するためのリソースを推奨します。これらの強力なツールは、非正規ゲノム領域における推定マイクロタンパク質コード配列の同定に役立てたり、目的の非コード転写産物中に翻訳可能性を有する保存されたコード配列の存在を排除するために使用できます。
Introduction
ゲノム中のコード要素の完全なセットの同定は、ヒトゲノムプロジェクトの開始以来、主要な目標であり、生物学的システムおよび遺伝ベースの疾患の病因の理解に向けた中心的な目的であり続けている1,2,3,4。NGS技術の進歩により、脊椎動物、無脊椎動物、酵母、植物など、広範な数の生物の全ゲノム配列が生産されました5。さらに、ハイスループット転写シーケンシング法は、細胞トランスクリプトームの複雑さをさらに明らかにし、タンパク質コード機能と非コード機能の両方を有する何千もの新規RNA分子を同定した6,7。この膨大な量の配列情報を解読することは進行中のプロセスであり、包括的な遺伝子アノテーションの取り組みには課題が残っています8。
リボソームプロファイリング9,10およびポリリボソームシーケンシング11を含む翻訳プロファイリング方法の最近の開発は、何百もの非正規翻訳事象がゲノム全体の現在注釈のないsORFにマッピングされ、マイクロタンパク質またはマイクロペプチド12,13,14,15,16と呼ばれる小さなタンパク質を生成する可能性を示す証拠を提供し、マイクロタンパク質は、そのサイズが小さく(<100アミノ酸)、古典的タンパク質コード遺伝子特性の欠如のために、以前は標準的な遺伝子注釈法によって見過ごされていた汎用性の高いタンパク質の新規クラスとして浮上している8,12,18,19,20。マイクロタンパク質は、酵母21、22、ハエ17、23、24、および哺乳類25、26、27、28を含む事実上すべての生物において記載されており、発生、代謝、およびストレスシグナル伝達を含む多様なプロセスにおいて重要な役割を果たすことが示されている19、20、29、 30,31,32,33,34。したがって、この長い間見過ごされてきた機能的な小さなタンパク質のクラスの追加のメンバーのゲノムを採掘し続けることが不可欠です。
マイクロタンパク質の生物学的重要性が広く認識されているにもかかわらず、このクラスの遺伝子はゲノム注釈において非常に過小評価されており、それらの正確な同定は、この分野の進歩を妨げている継続的な課題であり続けている。マイクロタンパク質コード配列の同定に関連する困難を克服するために、様々な計算ツールおよび実験方法が最近開発されている(いくつかの包括的なレビュー8,35,36,37で広く議論されている)。最近の多くのマイクロタンパク質同定研究38,39,40,41,42,43,44,45,46,47は、PhyloCSF 48,49と呼ばれるそのようなアルゴリズムの使用に大きく依存しているゲノムの保存されたタンパク質コード領域と非コード領域を区別するために利用できる強力な比較ゲノミクスアプローチです。
PhyloCSFは、多種ヌクレオチドアラインメントと系統発生モデルを使用してコドン置換頻度(CSF)を比較し、タンパク質コード遺伝子の進化的シグネチャを検出します。この経験的モデルベースのアプローチは、タンパク質が主にヌクレオチド配列ではなくアミノ酸レベルで保存されるという前提に依存している。したがって、同じアミノ酸をコードする同義のコドン置換、または保存された特性(すなわち、電荷、疎水性、極性)を有するアミノ酸へのコドン置換は正にスコア付けされ、ミスセンスおよびナンセンス置換を含む非同義置換は負にスコア付けされる。PhyloCSFは全ゲノムデータに基づいて訓練されており、マイクロタンパク質または標準タンパク質コード遺伝子の個々のエクソンを分析する際に必要な、コード配列(CDS)の短い部分を全配列から分離してスコアリングするのに有効であることが証明されています48,49。
特に、カリフォルニア大学サンタクルーズ校(UCSC)ゲノムブラウザ49,50,51にPhyloCSFトラックハブが最近統合され、あらゆるバックグラウンドの研究者がユーザーフレンドリーなインターフェースに簡単にアクセスして、関心のあるゲノム領域にタンパク質コーディングの可能性を問い合わせることができます。以下に概説するプロトコルは、UCSCゲノムブラウザにPhyloCSFトラックハブをロードし、その後、関心のあるゲノム領域を問い合わせて、信頼性の高いタンパク質コード領域(またはその欠如)をプローブする方法に関する詳細な指示を提供します。さらに、正のPhyloCSFスコアが観察される場合には、マイクロタンパク質コード電位をさらに分析し、同定されたアミノ酸配列の複数の種アラインメントを効率的に生成して、種間配列保存を例示するためのステップが描かれる。最後に、予測されたドメイン構造や推定マイクロタンパク質機能への洞察など、同定されたマイクロタンパク質特性を調査するために、いくつかの追加の公的に利用可能なリソースとツールが議論で紹介されています。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
以下に概説するプロトコルは、UCSCゲノムブラウザ(Mudgeらによって生成された)上のPhyloCSFブラウザトラックをロードしてナビゲートする手順を詳述している。UCSCゲノムブラウザに関する一般的な質問については、広範なゲノムブラウザユーザーズガイドがここにあります: https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html.
1. PhyloCSF トラックハブの UCSC ゲノムブラウザへのロード
- インターネットブラウザウィンドウを開き、UCSCゲノムブラウザ(https://genome.ucsc.edu/)に移動します。
- [ ツール ] 見出しで、[ ハブの追跡] オプションを選択します。
メモ: [ハブの追跡 ] オプションは、[ マイ データ ] タブの下にもあります。 - [パブリック ハブ] タブで、[検索用語] ボックスに「PhyloCSF」と入力します。[パブリックハブの検索]ボタンをクリックします。
- ハブ名PhyloCSF(説明:PhyloCSFによって測定される進化的タンパク質コード電位)の 接続 ボタンをクリックして、 PhyloCSF に接続します。
メモ: このトラックハブは、人間 (hg19 および hg38) およびマウス (mm10 および mm39) を含む多数のアセンブリにロードされます。 - 接続をクリックした後、UCSCゲノムブラウザゲートウェイページ(https://genome.ucsc.edu/cgi-bin/hgGateway)にリダイレクトされるのを待ちます。
2. 遺伝子識別子を使用した目的の遺伝子へのナビゲート
- 照会する種とゲノムアセンブリを選択します。別の種(マウスなど)を照会するには、適切なアイコンをクリックして [種の参照/選択] 見出しの下にある目的の種を選択するか、種、 共通名、またはアセンブリIDを入力してください。
メモ: アセンブリは、「 位置を検索」(Find Position ) 見出しのすぐ下にリストされています。通常、デフォルトはヒューマンアセンブリです(例:2009年12月[GRCh37/hg19])。 - ドロップダウンメニューを使用して 「位置を検索」(Find Position) 見出しで検索するアセンブリを選択します。
- 位置/検索用語ボックスに位置、遺伝子記号、または 検索用語 を入力し、「 移動 」をクリックしてゲノムブラウザ上の目的の遺伝子に移動します。
- 検索の結果、複数の一致が発生した場合は、関心のある位置の選択が必要なページにリダイレクトされるのを待ちます。 目的の適切な遺伝子をクリックします。
3. 配列情報を用いたゲノム関心領域への移動
- UCSCゲノムブラウザ(https://genome.ucsc.edu/)に移動し、当社のツール見出しの下にあるBLASTライクアライメントツール(BLAT)を選択して、特定のDNAまたはタンパク質配列を照会します。または、[ツール] タブの上にカーソルを置き、[Blat] オプションを選択するか、次のリンクをたどります: https://genome.ucsc.edu/cgi-bin/hgBlat。
- ドロップダウンメニューを使用して、目的の種(ゲノム)と アセンブリ を選択します。
- ドロップダウンメニューを使用して クエリタイプ を定義します。
- 目的のシーケンスを BLAT 検索ゲノム テキスト ボックスに貼り付け、[ 送信] をクリックします。
- 「アクション」見出しの下にあるブラウザー・リンクをクリックして、関心のあるゲノム領域に移動します。
4. PhyloCSFトラックデータを用いた保存されたsORFの同定
- 関心のあるゲノム領域を視覚的にスキャンして、PhyloCSF領域を肯定的にスコアリングします(図1)。
注:UCSCゲノムブラウザでPhyloCSFスコアを視覚的に解釈する方法の詳細については、以下の代表的な結果のセクションを参照してください。 - ズーム機能を使用して、関心領域を拡大し、配列特性を調べ、開始/停止コドンを検索します。手動でズームインするには、Shift キーを押しながらマウスボタンをクリックしたまま、関心領域に沿ってドラッグします。または、ページ上部のズームインボタンとズームアウトボタンを使用してナビゲートします(1.5倍、3倍、10倍、または基本ズームオプションを使用できます)。
注:ズーム イン/ズームアウト ボタンを使用する前に、関心領域が画面の中央になるように遺伝子の位置を変更する必要があります。この操作を実行するには、画像をクリックして左または右にドラッグし、必要に応じてゲノム領域を水平に移動するか、ページ上部の 移動 矢印を使用します。 - ヌクレオチド(塩基)配列が見えるまでズームインします。
注:ヌクレオチド配列は、+1平滑化フィロCSFスコアのすぐ上に表示されます。 - 正にスコアリングされたPhyloCSF領域の最初と最後付近の塩基配列を目視でスキャンし、推定開始(ATG)および停止(TGA/TAA/TAG)コドンを同定する。
注:目的の遺伝子がDNAのマイナス鎖上にある場合、開始コドンと停止コドンは逆補体になります(すなわち、開始コドンのCATと終止コドンのTCA/TTA/CTA)。
5. 他のゲノム中の相同領域を見る
- ページ上部の [表示 ]見出しの上にマウスを置き、[ 他のゲノム(変換)] オプションをクリックします。
- [新しいゲノム] 見出しの下にあるドロップダウン メニューを使用して、目的の ゲノム を定義します。
- 「新規アセンブリ」(New Assembly) 見出しの下のドロップダウンメニューを使用して目的のゲノム アセンブリ を選択し、「 送信」(Submit) ボタンをクリックします。
- ブラウザが類似性を持つ新しいアセンブリ内の領域のリストを返したら、 染色体位置 リンクをクリックして、関心のある相同領域に移動します。
注:全塩基(ヌクレオチド)の割合と領域によってカバーされるスパンは、リストされている領域ごとに定義されます。一致する塩基の割合が高いほど、関心領域の保存期間は高くなります。 - セクション 4 で説明したのと同じナビゲーション戦略に従って、シーケンスを分析します。
6. 目的のマイクロタンパク質の複数種配列アラインメントの生成
- UCSCゲノムブラウザのGENCODEトラック(図1Aに青いボックスで示されている)で目的の遺伝子をクリックして、遺伝子説明ページに移動します。
- 「シーケンス」および「ツールおよびデータベースへのリンク」見出しの下で、「他の種 FASTA」と表示される表のリンクをクリックします。
- 関心のある種に関連付けられているボックスをクリックして選択します。「 送信」をクリックします。ページの下部に表示されるシーケンスをコピーして、FASTA 形式でワープロ ドキュメントに貼り付けます。
- 2番目のブラウザウィンドウを開き、欧州バイオインフォマティクス研究所(EMBL-EBI)のウェブサイト53,54:https://www.ebi.ac.uk/Tools/msa/clustalo/ にあるClustal Omega Multiple Sequence Alignment tool52に移動します。
- クリップボードに残っているシーケンスファイルを、サポートされている任意のフォーマットでシーケンスを読み取る STEP 1 のボックスに貼り付けます。ページの一番下までスクロールし、[送信]をクリックします。各アミノ酸の保存の程度を示す記号(記号は表1に定義されている)について、整列された結果(黒いフォント)の下を見てください。
メモ: アライメントの生成には数分かかる場合があります。 - アミノ酸の特性を色で表示するには、シーケンスのすぐ上にある [色の表示] リンクをクリックして、その特性( 表2で定義)に従ってアミノ酸に色を付けます。
- シーケンスの配置をコピーしてワープロまたはスライドショープログラムに貼り付けて、図またはイラストファイル(図 2など)を生成します。
メモ: 整列には等幅フォントを使用します (Courier)。 - Clustal Omega の結果ページから他の出力を表示するには、適切なタブ (ガイドツリーまたは系統樹) をクリックします。
- 「結果ビューアラー」タブをクリックすると、複数の配列アライメント編集、視覚化、および分析55を専門とする無料プログラムであるJalviewを使用して配列情報を表示したり、MViewおよびSimple Phylogeny56への直接リンクにアクセスしたりできます。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
ここでは、検証済みのマイクロタンパク質ミトレグリン(Mtln)を例として使用して、保存されたsORFがUCSCゲノムブラウザで簡単に視覚化および分析できる正のPhyloCSFスコアを生成する方法を実証します。ミトレグリンは、以前は非コードRNAとして注釈が付けられていた(旧ヒト遺伝子ID LINC00116およびマウス遺伝子ID 1500011K16Rik)。 比較ゲノミクスおよび配列保存解析法は、その最初の発見40、57、58、59、60、61において重要な役割を果たし、これらの方法の強さを強調した。この例では、マウス GRCm38/mm10 (2011 年 12 月) アセンブリが使用されます。検索は、プロトコルセクション2に記載されているように、遺伝子識別子(ミトレグリン、Mtln)または遺伝子位置(chr2:127,791,364-127,792,496)を使用して行うことができる。あるいは、ミトレグリンのアミノ酸配列(図2に示す)は、BLATツール(プロトコルセクション3に記載)を用いて検索することができる。
図 1A に示すような画面が表示され、画面の上部に PhyloCSF トラックハブが表示されます。平滑化されたPhyloCSFトラック(各コドンがコード化している確率を定義する隠れマルコフモデルで平滑化)は、DNAのプラス鎖に対応する3つのトラック(緑色でPhyloCSF +1、+2および+3として描かれる)およびDNAのマイナス鎖に対応する3つのトラック(赤色でPhyloCSF -1として描かれる)を有する6つの合計トラックとして描かれ、 -2 および -3)。これらのトラックは、各方向における目的の遺伝子の3つの潜在的なリーディングフレームを表す。ブラウザウィンドウでは、エクソンはイントロンを表す細い青い水平線で接続された青い長方形として描かれます。イントロニック領域の矢印は、遺伝子がどの方向に転写されるか(したがって、PhyloCSFスコアのためにどの鎖に焦点を当てるべきか)を示す。図 1 の Mtln の例では、イントロニック矢印が左を指しています。したがって、Mtln遺伝子はDNAのマイナス鎖から転写され、関連するPhyloCSFスコアは-1、-2、および-3トラック(赤色)に示されます。
各PhyloCSFトラックは細い黒い線で描かれ、負のスコアリング領域は線の下に明るい緑/赤で示され、正のスコアリング領域は線の上に濃い緑/赤で示されます。序文で説明したように、正のPhyloCSFスコアは、コーディングされている可能性が高い保存領域を示します。特に高い配列保存を有するタンパク質コード領域については、アンチセンス鎖上でも正のスコアを付けることが多いことに留意されたい。しかし、PhyloCSFスコアは通常、正しい鎖の方が高い。例えば、これは、正しいコード配列がPhyloCSF -1トラックにおいて非常に高くスコア付けされ、アンチセンス鎖(PhyloCSF +2トラック)も正のスコアを生成するMtlnの図1で見ることができる。図1A(ブラックボックスで示す)に見られるように、Mtlnの最初のエクソンには、PhyloCSF−1トラック上で非常に高いスコアを獲得する領域があり、これはコーディング領域に対応する可能性があることを示唆している。この領域をさらに詳しく調べるには、領域を拡大して拡大すると便利です(図1B)。図1C,Dに示すように、Mtlnの最初のエクソンにおける正のスコアリング領域は、開始コドン(図1C)の真上で始まり、終止コドン(図1D)で終わるが、これは、このORFが高度に保存されていることを示し、コードORFであることを強く示唆する。MtlnはDNAのマイナス鎖上にあるため、開始コドンと停止コドンはコドンの逆補体として示される(すなわち、ATG開始コドンはCATとして示され[図1C]、TGA停止コドンはTCA[図1D]として示される)。
この技術は、PhyloCSFを使用してマイクロタンパク質コード電位を有する保存領域を探索することに加えて、保存されたORFの存在を除外する推定非コードRNAのファーストパス分析としても適用することができ、したがって非コード注釈のサポートを提供する。例えば、PhyloCSFを用いた十分に特徴付けられたlncRNA HOTAIR62,63の解析は、6つのトラックすべてにわたって遺伝子全体にわたって負のスコアを示し(図3)、配列保存の欠如を強く示し、HOTAIRが非コードRNAとして正しく注釈付けされていることを裏付ける。
図1に明確に見られるように、ミトレグリンのコーディングORF全体が単一のエクソン内に位置するため、PhyloCSFは、中断のない単一の正のスコアリング領域を持つシンプルで簡単な読み出しを生成します。しかし、PhyloCSFトラックハブデータは、必ずしも明確で解釈しやすいとは限りません。例えば、マウス1810058I24Rik遺伝子47,64,65によってコードされるミトランバン/Stmp1/Mm47マイクロタンパク質は、3つのエクソンにまたがる保存されたORFを描いており(図4A)、正のPhyloCSFスコアはエクソン1の+2トラック(図4B)からエクソン2の+3トラック(図4C)にジャンプし、その後エクソン3の+2トラックに戻る(図4D)。).一見すると混乱しているように見えますが、説明は非常に簡単です。PhyloCSFは、各遺伝子の特定のエクソン/イントロンアーキテクチャを考慮せずに、ゲノム領域の6つの潜在的なリーディングフレーム(DNAのプラス鎖に3つ、マイナス鎖に3つ)をスコアリングします。そのため、リーディングフレームの3塩基周期性におけるイントロニック配列情報を保持している。したがって、イントロンに3つで割り切れないヌクレオチド(すなわち、3つのヌクレオチド/コドン)が多数含まれている場合、PhyloCSFリーディングフレームはあるトラックから別のトラックにジャンプします。
最後に、PhyloCSFは、単一のRNA分子内の複数の異なるコードORFを同定するためにも効果的に使用することができる。例えば、MIEF1マイクロタンパク質(MIEF1-MP)は、ミトコンドリア伸長因子1(MIEF1)66の5'UTR内にコードされる(図5)。MIEF1ゲノム領域をPhyloCSFによって解析すると、MIEF1-MP(図5C)に対応する離散的な正のPhyloCSFスコアが、MIEF1のメインCDSの上流で容易に観察され得る(図5B)。MIEF1およびそれに関連するマイクロタンパク質(MIEF1-MP)に関するさらなる議論は、この記事で概説されている方法およびプロトコルの長所と短所の要約とともに、議論の中で以下に提供される。

図1:ミトレグリン(Mtln)遺伝子の系統CSF解析は、検証されたマイクロタンパク質に対応する高い配列保存の領域を示す。 (A) UCSC Genome Browser と PhyloCSF Tracks のスクリーンショットは、Mtln に 2 つのエクソンと 1 つのイントロンが含まれていることを示している。イントロン内の矢印は左を指し、Mtln遺伝子がDNAのマイナス鎖から転写されたことを示し、したがって、関連するPhyloCSFスコアは-1、-2、および-3トラック(赤色)に示されている。完全なミトレグリンコード配列はエクソン1内に含まれ、PhyloCSF-1トラック(B)で高いスコアを獲得します。保存された開始コドンは、PhyloCSF−1トラック(C)における正のスコアリング領域の先頭で明確に観察することができ、緑色のボックス(CAT、逆補体ATG)で強調表示される。さらに、保存された終止コドン(TCA、逆補体TGA)は、パネル(D)に赤いボックスで示され、これは正にスコアリングされたPhyloCSF領域の末端と一致する。Mtln遺伝子に関する詳細情報は、青いボックス(パネルAに示されている)内のMtln遺伝子識別子をクリックすることで見つけることができます。注目すべきは、高度に保存されたタンパク質コード領域は、アンチセンス鎖でも陽性のスコアを示すことが多い(MtlnのPhyloCSF +2トラックで見られる)。ただし、PhyloCSFスコアは通常、正しい鎖(この例ではPhyloCSF-1トラック)で高くなります。この図の拡大版を表示するには、ここをクリックしてください。

図2:Clustal Omegaプログラムを用いて生成された微小タンパク質ミトレグリンの複数種配列アラインメント。 示された8種のミトレグリンアミノ酸配列を、プロトコルセクション6に詳述されているように抽出し、Clustal Ω多重配列アラインメントツールとアラインメントした。アミノ酸の特性は、色(赤、小/疎水性、青、酸性、マゼンタ、塩基性、緑、ヒドロxl/スルフヒドリル/アミン)( 表2でさらに定義)で示されます。アミノ酸の下の記号は、保存の程度(アスタリスク、完全に保存された残基;結腸、強く類似した特性を有するアミノ酸;期間、弱く類似した特性のグループ間の保存)を示す( 表1に広く詳述されている)。 この図の拡大版を表示するには、ここをクリックしてください。
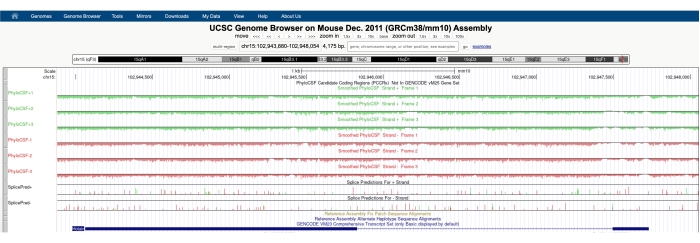
図3:検証済みの長いノンコーディングRNA HotairのPhyloCSFトラックのスクリーンショットは、そのゲノム遺伝子座全体に配列保存の欠如を示しています。Hotair のイントロニック領域の矢印は左を向いており、lncRNA が DNA の負鎖から転写されていることを示しているため、PhyloCSF -1、-2、および -3 トラックが解析の焦点となるはずです。PhyloCSFスコアは遺伝子全体(6つのトラックすべて)で負であり、配列保存の欠如を示しており、ノンコーディングRNAとしての適切な注釈を支持していることに注意してください。この図の拡大版を表示するには、ここをクリックしてください。

図4:マイクロタンパク質mitolamban/Stmp1/Mm47をコードするマウス1810058I24Rik遺伝子の系統CSF解析(A)マウス1810058I24Rik遺伝子は3つのエクソンで構成されており、イントロニック領域の矢印は右を指しており、DNAのプラス鎖に転写されていることを示しているため、PhyloCSF +1、+2、および+3トラックを分析する必要があります。 保存されたマイクロタンパク質コード配列は、エクソン1(B)から始まり、エクソン2(C)を通って読み取り、そしてエクソン3(D)で終わる3つのエクソンすべてにまたがる。正の PhyloCSF スコアは、エクソン 1 の +2 トラック、エクソン 2 の +3 トラック、エクソン 1 の +2 トラックにあることに注意してください。あるトラックから別のトラックに正のスコアが移動する理由は、PhyloCSFが遺伝子のエクソン/イントロン構造とは無関係にDNA配列の6つの潜在的なリーディングフレームを分析するためです。したがって、3つ(3つのヌクレオチド/コドン)で割り切れない多数のヌクレオチドを含むイントロンは、リーディングフレームを別のトラックにシフトさせる。この図の拡大版を表示するには、ここをクリックしてください。

図5: Mief1 ゲノム遺伝子座をPhyloCSFで解析したところ、5' UTRにおいて、共有RNA上のメインMief1 CDSから独立したタンパク質コード電位を有する領域が同定された。 この保存された上流ORF(uORF)は、Mief1−MPという名前の微量タンパク質をコードすることが示されている。(A) Mief1 ゲノム遺伝子座の概要。イントロンの矢印は右を指しており、Mief1がDNAのプラス鎖から転写されていることを示しています(コーディング電位を決定するためにPhyloCSF +1、+2、および+3トラックに焦点を当てます)。メインのMief1 CDSは、463アミノ酸タンパク質をコードし、パネル(B)に示されている。しかし、Mief1の5'UTR内には、Mief1-MP(C)と呼ばれるユニークな70アミノ酸マイクロタンパク質をコードする明確な保存された上流ORFもあります。パネル Cに見られるように、Mief1-MPはMief1 5' UTR内に独自の保存された開始コドンと停止コドンを持ち、ORFはPhyloCSF +1トラックで非常に高く評価され、機能的なマイクロタンパク質をコードするという強力な証拠を提供します。略語: ORF = オープンリーディングフレーム;uORF = アップストリーム ORF;UTR = 非翻訳領域;CDS = 符号化配列。 この図の拡大版を表示するには、ここをクリックしてください。
| 記号 | アミノ酸保存レベル | グループ化されたアミノ酸 |
| アスタリスク (*) | 完全に保存された残留物 | 該当なし(単一、完全保存残留物) |
| コロン (:) | プロパティが強く類似しているグループ | スタ;ネクク;NHQK;NDEQ;QHRK;ミルブ;ミルフ;ハイ;2009年度 |
| ピリオド (.) | 類似性が弱いグループ | CSA;ATV;サグ;ティッカーSTPA;SGND;SNDEQK;NDEQHK;ネクハルク;FVLIM;ティッカー |
| スペース (記号なし) | 類似点なし | 該当なし (類似性なし) |
表1:Clustal Omegaによって生成された複数のシーケンスアラインメントのコンセンサスシンボルの定義。 図2に示す複数種の配列アラインメントは、Clustal Omega52を使用して生成した。略語:セリン(S)、スレオニン(T)、アラニン(A)、アスパラギン(N)、グルタミン酸(E)、グルタミン(Q)、リジン(K)、アスパラギン酸(D)、アルギニン(R)、メチオニン(M)、イソロイシン(I)、ロイシン(L)、フェニルアラニン(F)、ヒスチジン(H)、チロシン(Y)、トリプトファン(W)、システイン(C)、バリン(V)、グリシン(G)、プロリン(P)。
| フォントの色 | 財産 | アミノ酸残基 [略称] |
| 赤い | 小型、疎水性 | アラニン[A]、バリン[V]、フェニルアラニン[F]、プロリン[P]、メチオニン[M]、イソロイシン[I]、ロイシン[L]、トリプトファン[W] |
| 青い | 酸性 | アスパラギン酸[D]、グルタミン酸[E] |
| マジェンタ | 基本的な | アルギニン[R]、リジン[K] |
| 緑 | ヒドロキセル、スルフヒドリル、アミン、+G | セリン[S]、スレオニン[T]、チロシン[Y]、ヒスチジン[H]、システイン[C]、アスパラギン[N]、グリシン[G]、グルタミン[Q] |
表2: 図2に描かれたアミノ酸の特性。 Clustal Ω52 は、 図2に示す多重配列アラインメントを生成するために使用した。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
ここで紹介するプロトコルは、ユーザーフレンドリーなUCSCゲノムブラウザ48,49,50,51上のPhyloCSFを使用して、マイクロタンパク質コードの可能性について関心のあるゲノム領域を問い合わせる方法に関する詳細な指示を提供します。上記で詳述したように、PhyloCSFは、系統発生モデルとコドン置換頻度を統合して、タンパク質コード遺伝子の典型的な進化的シグネチャを同定する強力な比較ゲノミクスアルゴリズムである48,49。 PhyloCSFは、以前に非コード38、39、40、41、42、43、44、45、46、47として注釈が付けられたゲノム領域における機能的マイクロタンパク質を同定するために広く使用されている。そして、このアプローチは、13アミノ酸の小さなマイクロタンパク質や正準タンパク質の小さなエクソンなどの短い配列について、他の比較ゲノミクス法よりも優れていることが示されています35、48、49。特に、進化的保存を通じて機能的タンパク質コード配列を同定する堅牢な方法としてのPhyloCSFの有用性は、脊椎動物および無脊椎動物の種のそれを超えて広がり、SARS-CoV-2ゲノムのタンパク質コード能力の調査に成功するためにウイルスゲノムに最近適用された67。
PhyloCSFの利点は、注釈付き非コードRNA内の推定コード配列を同定することに加えて、5'上流および3'下流の両方のORF(それぞれuORFおよびdORF)を含む、正準タンパク質コード遺伝子の注釈付き非翻訳領域(UTR)内のORFによってコードされる保存されたマイクロタンパク質を確実に検出できることである8,19,66,68 .例えば、MIEF1マイクロタンパク質(MIEF1−MP)は、ミトコンドリア伸長因子1(MIEF1)66の5'UTRにコードされる。MIEF1-MPの場合、MIEF1-MPに対応する離散的な正のPhyloCSFスコアが、MIEF1をコードするORFの上流で観察される(図5)。いくつかのuORFコードマイクロタンパク質は、それらの共有mRNA上の下流の正準タンパク質(例:MIEF1-MPおよびMIEF1)と直接相互作用するが、他のものはメインCDS66,68によってコードされるタンパク質とは独立して機能する。したがって、uORFコードされたマイクロタンパク質を特徴付けるとき、それらがそれらの下流タンパク質産物の直接調節を介して機能すると仮定されるべきではない。
PhyloCSFは、保存されたマイクロタンパク質コード配列を同定するためのツールとして多くの明確な強みを持っていますが、この方法のいくつかの限界を認識することが重要です。第一に、配列保存は、ゲノム領域が機能的選択を受け、したがってコーディングされていることを強く示唆しているが、堅牢な保存の欠如および結果として生じる負のPhyloCSFスコアは、所与の配列のコード可能性を決定的に排除するものではない。言い換えれば、PhyloCSFのみに依存することは、強く保存されていないが機能的なマイクロタンパク質を産生する翻訳ORFの監督をもたらす可能性がある。特に、保存スコアが低い、または保存スコアが負のゲノム領域は、種特異的コード領域または配列発散またはde novo遺伝子誕生46,69,70,71,72,73,74を介した進化的「若い」遺伝子のコード領域に対応する可能性がある。例えば、以前はヒト非コードRNA LINC00467であると考えられていたものによってコードされるマイクロタンパク質ASAPは、アミノ酸配列が高等哺乳動物においてのみ保存されているため、PhyloCSFによって正のスコアリングを受けていない75。さらに、最近の研究では、遺伝子間lncRNA RP3-527G5.1によってコードされるものを含む、PhyloCSFスコア68,72の正をもたらさないいくつかのヒト特異的マイクロタンパク質が同定された。この点で、正のPhyloCSFスコアの欠如は、非コード領域の証拠として解釈することができず、注意して解釈されるべきである。
PhyloCSFを使用する際に留意すべき2番目の考慮事項は、正のスコアが機能選択とタンパク質コード能力を非常に示唆するものであるにもかかわらず、この一連の証拠は単独では立たず、実験的に検証されなければならないということです。安定したマイクロタンパク質発現の裏付けとなる証拠を生成するために使用できる方法の例には、目的のマイクロタンパク質配列に対して惹起された抗体を用いた質量分析またはウェスタンブロッティングによる推定タンパク質の検出が含まれる。あるいは、最適な抗原性のための配列選択がないため、マイクロタンパク質に対する信頼性の高い抗体を作製することは困難であり得るため、CRISPR/Cas9および相同性指向修復(HDR)経路を使用して、推定されるマイクロタンパク質配列とインフレームで内在性遺伝子座にエピトープタグを導入し、それによって高親和性抗体を用いた目的のタンパク質の検出を容易にすることもできる(例えば、 フラグ、HA、V5、Myc)18。PhyloCSFが認める最後の制限は、 現在、ホモサピエンス (ヒトhg19、hg38)、 Mus musculus (マウスmm10、mm39)、 Gallus gallus (ニワトリ、galGal4、galGal6)、 ショウジョウバエメラノガスター (フルーツフライ、dm6)、 Caenorhabditis elegans など、一般的に使用されているゲノムアセンブリの多くに統合されているということです。 (線虫、ce11)、およびSARS-CoV-2(wuhCor1)には、現在UCSCゲノムブラウザで直接照会できない種がまだたくさんあります。
同定されたマイクロタンパク質内の保存ドメインまたは配列特性の同定は、それらの機能的関連性に対する信頼性を高め、それらの推定機能に対するいくつかの洞察を提供するのに役立つ。ここでは、同定されたマイクロタンパク質アミノ酸配列をさらに詳細に分析してそのような洞察を得るために使用できる特定のツールとリソースに関する推奨事項を提供します。以下にリストされている特定のツール(および材料表に要約されている)は一般に自由に利用可能であり、我々はそれらがマイクロタンパク質研究18,38,39,40,41,47において特にユーザーフレンドリーで堅牢であることを発見しました。ここで説明するツール以外にも、Expasy(https://www.expasy.org)やEMBL-EBI(https://www.ebi.ac.uk/services/all)などのバイオインフォマティクスリソースポータルには、他にも多数のリソースがあります。ただし、これらのリポジトリ内の各ツールの詳細を詳述することは、この記事の範囲外です。ここでは、次のリソースをお勧めします。
まず、TMHMM76 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0)は、膜貫通ドメインの存在について目的のタンパク質配列を分析する。特に、これまで機能的に特徴付けられてきた多くのマイクロタンパク質は、シングルパス膜貫通ドメインを含み、これは膜領域への局在化を容易にし、イオンチャネル、交換体、および膜関連酵素の直接的な調節を可能にする30。第二に、National Center for Biotechnology Information(NCBI)Conserved Domain Search77 (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)は、タンパク質またはコードヌクレオチド配列内の保存ドメインを同定するために使用される一般的なツールです。第三に、タンパク質ファミリー(Pfam)78 データベース(http://pfam.xfam.org)は、タンパク質ファミリーおよびドメインのアラインメントおよび分類を提供する。第四に、WoLF PSORT79 (https://wolfpsort.hgc.jp/)は、細胞内タンパク質の局在を予測するために使用することができるツールである。第五に、COXPRESdB80 は、遺伝子機能を推定するために共調節された遺伝子関係を提供する遺伝子共発現データベース(https://coxpresdb.jp)である。最後に、SignalP 6.081 は、シグナルペプチド配列の存在を認識し、切断部位の位置を予測する、広く使用されている予測プログラム(https://services.healthtech.dtu.dk/service.php?SignalP)である。
要約すると、ここで説明する方法は、UCSCゲノムブラウザ上のPhyloCSFを使用して、タンパク質コード電位について関心のあるゲノム領域を効果的に分析するために使用することができる。これらの方法はアクセス性が高く、バイオインフォマティクスや比較ゲノミクスの事前の訓練や専門知識がなくても、個人が簡単に習得し、効率的に適用することができます。ここで詳細に示されているように、PhyloCSFは、脊椎動物、無脊椎動物、およびウイルスゲノムにおけるタンパク質コード遺伝子と非コード遺伝子を区別するのに役立つファーストパス解析として適用できる強力なツールであり、このアプローチの長所は指摘された弱点をはるかに上回ります。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者らは、競合する金銭的利益はないと宣言している。
Acknowledgments
この研究は、国立衛生研究所(HL-141630およびHL-160569)とシンシナティ小児研究財団(受託者賞)からの助成金によって支援されました。
Materials
| Name | Company | Catalog Number | Comments |
| Website | Website Address | Requirements | |
| Clustal Omega Multiple Sequence Alignment Tool | https://www.ebi.ac.uk/Tools/msa/clustalo/ | Web browser | Multiple sequence alignment program for the efficient alignment of FASTA sequences (i.e. for cross-species comparison of identified microproteins) |
| COXPRESSdb | https://coxpresdb.jp | Web browser | Provides co-regulated gene relationships to estimate gene functions |
| EMBL-EBI Bioinformatics Tools FAQs | https://www.ebi.ac.uk/seqdb/confluence/display/JDSAT/Bioinformatics+Tools+FAQ | Web browser | Frequently Asked Questions (FAQs) for EMBL-EBI tools. Includes the color coding key for protein sequence alignments |
| European Bioinformatics Institute (EMBL-EBI), Tools and Data Resources |
https://www.ebi.ac.uk/services/all | Web browser | Comprehensive list of freely available websites, tools and data resources |
| Expasy - Swiss Bioinformatics Resource Portal | https://www.expasy.org | Web browser | Suite of bioinformatic tools and resources for protein sequence analysis that is maintained by the Swiss Institute of Bioinformatics (SIB) |
| National Center for Biotechnology Information (NCBI) Conserved Domain Search |
https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi | Web browser | Search tool to identify conserved domains within protein or coding nucleotide sequences |
| Pfam 35 | http://pfam.xfam.org | Web browser | Protein family (Pfam) database, provides alignments and classification of protein families and domains |
| PhyloCSF Track Hub Description | https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1267045267_TEc99h2oW5Q edaCd4ir8aZ65ryaD&db=mm10 &c=chr2&g=hub_109801_ PhyloCSF_smooth |
Web browser | Detailed description of the Smoothed PhyloCSF tracks and PhyloCSF Track Hub |
| SignalP 6.0 | https://services.healthtech.dtu.dk/service.php?SignalP-6.0 | Web browser | Predicts the presence of signal peptides and the location of their cleavage sites |
| TMHMM - 2.0 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 | Web browser | Prediction of transmembrane helices in proteins |
| UCSC Genome Browser BLAT Search | https://genome.ucsc.edu/cgi-bin/hgBlat | Web browser | Tool used to find genomic regions using DNA or protein sequence information |
| UCSC Genome Browser Gateway | https://genome.ucsc.edu/cgi-bin/hgGateway | Web browser | Direct link to the UCSC Genome Browser Gateway |
| UCSC Genome Browser Home | https://genome.ucsc.edu/ | Web browser | Home website for the UCSC Genome Browser |
| UCSC Genome Browser Track Data Hubs | https://genome.ucsc.edu/cgi-bin/hgHubConnect#publicHubs | Web browser | Direct link to Track Data Hubs/Public Hubs database to search for and load the PhyloCSF Tracks |
| UCSC Genome Browser User Guide | https://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html | Web browser | Comprehensive user guide detailing how to navigate the UCSC Genome Browser |
| WoLF PSORT | https://wolfpsort.hgc.jp | Web browser | Protein subcellular localization prediction tool |
References
- Collins, F. S., Morgan, M., Patrinos, A. The human genome project: lessons from large-scale biology. Science. 300 (5617), 286-290 (2003).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Sachidanandam, R., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409 (6822), 928-933 (2001).
- Venter, J. C., et al.
- Fuentes-Pardo, A. P., Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular Ecology. 26 (20), 5369-5406 (2017).
- Carninci, P., et al. The transcriptional landscape of the mammalian genome. Science. 309 (5740), 1559-1563 (2005).
- Maeda, N., et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2 (4), 62 (2006).
- Schlesinger, D., Elsasser, S. J. Revisiting sORFs: overcoming challenges to identify and characterize functional microproteins. The FEBS Journal. 289 (1), 53-74 (2022).
- Ingolia, N. T., et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 8 (5), 1365-1379 (2014).
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324 (5924), 218-223 (2009).
- Aspden, J. L., et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 3, 03528 (2014).
- Andrews, S. J., Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nature Reviews Genetics. 15 (3), 193-204 (2014).
- Mackowiak, S. D., et al. Extensive identification and analysis of conserved small ORFs in animals. Genome Biology. 16 (1), 1-21 (2015).
- Ruiz-Orera, J., Messeguer, X., Subirana, J. A., Alba, M. M. Long non-coding RNAs as a source of new peptides. Elife. 3, 03523 (2014).
- Basrai, M. A., Hieter, P., Boeke, J. D. Small open reading frames: beautiful needles in the haystack. Genome Research. 7 (8), 768-771 (1997).
- Frith, M. C., et al. The abundance of short proteins in the mammalian proteome. PLoS Genetics. 2 (4), 52 (2006).
- Ladoukakis, E., Pereira, V., Magny, E. G., Eyre-Walker, A., Couso, J. P. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biology. 12 (11), 118 (2011).
- Makarewich, C. A., Olson, E. N.
- Wright, B. W., Yi, Z., Weissman, J. S., Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends in Cell Biology. , (2021).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Kastenmayer, J. P., et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 16 (3), 365-373 (2006).
- Smith, J. E., et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Reports. 7 (6), 1858-1866 (2014).
- Lin, M. F., et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 17 (12), 1823-1836 (2007).
- Magny, E. G., et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 341 (6150), 1116-1120 (2013).
- Bazzini, A. A., et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33 (9), 981-993 (2014).
- Ingolia, N. T., Lareau, L. F., Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147 (4), 789-802 (2011).
- Ma, J., et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 13 (3), 1757-1765 (2014).
- Slavoff, S. A., et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nature Chemical Biology. 9 (1), 59-64 (2013).
- Khitun, A., Ness, T. J., Slavoff, S. A. Small open reading frames and cellular stress responses. Molecular Omics. 15 (2), 108-116 (2019).
- Makarewich, C. A. The hidden world of membrane microproteins. Experimental Cell Research. 388 (2), 111853 (2020).
- Pueyo, J. I., Magny, E. G., Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends in Biochemical Sciences. 41 (8), 665-678 (2016).
- Pauli, A., et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 343 (6172), 1248636 (2014).
- Chng, S. C., Ho, L., Tian, J., Reversade, B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Developmental Cell. 27 (6), 672-680 (2013).
- Lee, C., et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism. 21 (3), 443-454 (2015).
- Pauli, A., Valen, E., Schier, A. F. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 37 (1), 103-112 (2015).
- Plaza, S., Menschaert, G., Payre, F. In search of lost small peptides. Annual Review of Cell and Developmental Biology. 33, 391-416 (2017).
- Kiniry, S. J., Michel, A. M., Baranov, P. V. Computational methods for ribosome profiling data analysis. Wiley Interdisciplinary Reviews: RNA. 11 (3), 1577 (2020).
- Anderson, D. M., et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 160 (4), 595-606 (2015).
- Anderson, D. M., et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Science Signaling. 9 (457), (2016).
- Makarewich, C. A., et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid beta-oxidation. Cell Reports. 23 (13), 3701-3709 (2018).
- Nelson, B. R., et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 351 (6270), 271-275 (2016).
- Chu, Q., et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat Commun. 10 (1), 4883 (2019).
- Senis, E., et al. TUNAR lncRNA encodes a microprotein that regulates neural differentiation and neurite formation by modulating calcium dynamics. Frontiers in Cell and Developmental Biology. 9, 747667 (2021).
- Li, M., et al. A putative long noncoding RNA-encoded micropeptide maintains cellular homeostasis in pancreatic beta cells. Molecular Therapy-Nucleic Acids. 26, 307-320 (2021).
- Martinez, T. F., et al. Accurate annotation of human protein-coding small open reading frames. Nature Chemical Biology. 16 (4), 458-468 (2020).
- van Heesch, S., et al. The translational landscape of the human heart. Cell. 178 (1), 242-260 (2019).
- Makarewich, C. A., et al. The cardiac-enriched microprotein mitolamban regulates mitochondrial respiratory complex assembly and function in mice. Proceedings of the National Academy of Sciences of the United States of America. 119 (6), 2120476119 (2022).
- Lin, M. F., Jungreis, I., Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 27 (13), 275-282 (2011).
- Mudge, J. M., et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Research. 29 (12), 2073-2087 (2019).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- Raney, B. J., et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 30 (7), 1003-1005 (2014).
- Sievers, F., et al. scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7 (1), 539 (2011).
- Goujon, M., et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research. 38 (2), 695-699 (2010).
- Harte, N., et al. Public web-based services from the European Bioinformatics Institute. Nucleic Acids Research. 32 (2), 3-9 (2004).
- Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., Barton, G. J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25 (9), 1189-1191 (2009).
- Madeira, F., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research. 47 (1), 636-641 (2019).
- Friesen, M., et al. Mitoregulin controls beta-oxidation in human and mouse adipocytes. Stem Cell Reports. 14 (4), 590-602 (2020).
- Stein, C. S., et al. Mitoregulin: A lncRNA-Encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Reports. 23 (13), 3710-3720 (2018).
- Chugunova, A., et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proceedings of the Nationall Academy of Sciences of the United States of America. 116 (11), 4940-4945 (2019).
- Lin, Y. F., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death and Disease. 10 (7), 528 (2019).
- Wang, L., et al. The micropeptide LEMP plays an evolutionarily conserved role in myogenesis. Cell Death and Disease. 11 (5), 357 (2020).
- He, S., Liu, S., Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evolutionary Biology. 11 (1), 1-14 (2011).
- Rinn, J. L., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129 (7), 1311-1323 (2007).
- Bhatta, A., et al. A Mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. Journal of Immunology. 204 (2), 428-437 (2020).
- Zhang, D., et al. Functional prediction and physiological characterization of a novel short trans-membrane protein 1 as a subunit of mitochondrial respiratory complexes. Physiological Genomics. 44 (23), 1133-1140 (2012).
- Rathore, A., et al.
- Jungreis, I., Sealfon, R., Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications. 12 (1), 2642 (2021).
- Chen, J., et al. Pervasive functional translation of noncanonical human open reading frames. Science. 367 (6482), 1140-1146 (2020).
- Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Canas, J. L., Messeguer, X., Alba, M. M. Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nature Ecology and Evolution. 2 (5), 890-896 (2018).
- Blevins, W. R., et al. Uncovering de novo gene birth in yeast using deep transcriptomics. Nature Communications. 12 (1), 604 (2021).
- Papadopoulos, C., et al. Intergenic ORFs as elementary structural modules of de novo gene birth and protein evolution. Genome Research. , (2021).
- Vakirlis, N., Duggan, K. M., McLysaght, A. De novo birth of functional, human-specific microproteins. bioRxiv. , 462744 (2021).
- Van Oss, S. B., Carvunis, A. R.
- Andersson, D. I., Jerlstrom-Hultqvist, J., Nasvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harbor Perspectives in Biology. 7 (6), 017996 (2015).
- Ge, Q., et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. Journal of Clinical Investigations. 131 (22), (2021).
- Sonnhammer, E. L., von Heijne, G., Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings. International Conference on Intelligent Syststems for Molecular Biology. 6, 175-182 (1998).
- Lu, S., et al.
- Mistry, J., et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 49, 412-419 (2021).
- Horton, P., et al.
- Obayashi, T., Kagaya, Y., Aoki, Y., Tadaka, S., Kinoshita, K. COXPRESdb v7: a gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Research. 47, 55-62 (2019).
- Teufel, F., et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology. , 01156 (2022).
