

Summary
Aquí nos demuestran los protocolos para la realización de una sola molécula de microscopía de fluorescencia en células vivas de las bacterias para permitir funcionales complejos moleculares para detectar, seguir y cuantificar.
Abstract
Una visión completa de los mecanismos de las células vivas sólo puede lograrse mediante la investigación de los procesos clave que provocan y dirigir los eventos a nivel celular. Hasta la fecha, la complejidad de corte de los sistemas biológicos ha provocado precisa una sola molécula de la experimentación a ser demasiado exigente, en lugar de centrarse en los estudios de sistemas de un solo uso a granel relativamente crudo media del conjunto de mediciones. Sin embargo, muchos procesos importantes ocurren en la célula viva en el ámbito de una o unas pocas moléculas, las mediciones del conjunto en general, la máscara de la naturaleza estocástica y heterogéneo de estos eventos. En este caso, mediante microscopía óptica avanzada y herramientas de análisis de análisis de imagen que se muestra cómo controlar las proteínas dentro de una sola célula viva bacteriana con una precisión de las moléculas individuales y cómo podemos observar la dinámica dentro de los complejos moleculares en el funcionamiento de máquinas biológicas. Las técnicas son directamente relevantes fisiológicamente. Son mínimamente perturbativa y no invasiva a la muestra biológica objeto de estudio y están totalmente adaptados para las investigaciones en la materia viva, las características no disponibles para los otros enfoques de una sola molécula de la biofísica. Además, las muestras biológicas estudiadas todos los productos de proteína fluorescente con etiquetas en niveles que son casi idénticas a las cepas de células no modificadas ("codificación genómica"), en comparación con el enfoque más común pero menos ideal para generar mucha más proteína que se producen de forma natural ('plásmido de expresión "). Por lo tanto, las muestras biológicas reales que serán investigados son significativamente más cerca de los organismos naturales, y por lo tanto, las observaciones más relevantes para los procesos fisiológicos reales.
Protocol
- Para iniciar este procedimiento, 50 l de material congelado de la proteína fluorescente expresando Escherichia coli células bacterianas primero se descongela y se cultiva aeróbicamente con agitación en 5 ml de medio de cultivo LB noche a 37 º C. Por la mañana, 50 l de esta cultura saturada se extrae y sub-cultivadas en un mínimo de M63 medios de glucosa en la cultura, la incubación a 30 º C durante 4 a 6 horas. Aquí se demuestra el uso de dos cepas diferentes de células, una de las cuales expresa un transporte de electrones del citocromo fusionado a GFP, y el otro que expresa una proteína implicada en el motor flagelar bacteriano fusionado a GFP.
- Células o bien puede recogidas directamente del crecimiento sub-cultura, si los ve como muestras inmovilizadas, o pueden ser cortados para truncar los flagelos bacterianos ver si en "atar" las condiciones.
- Esquila consiste en colocar por lo general 1-5 ml de la sub-cultura en un dispositivo que consta de dos jeringas estériles se unió por un tubo estéril. El corte se realiza por la alternancia de empujar en cada bomba de jeringa para impulsar la cultura a través del tubo estrecho. Esto se hace 50-100 veces, dependiendo de la extensión del corte requerido. La cultura se centrifuga para sedimentar las células, que se vuelve a suspender en medios mínimos para eliminar los fragmentos de los flagelos.
- A continuación, preparar limpiar BK7 cubreobjetos de vidrio por inmersión en una solución saturada de KOH en etanol durante 20 minutos, luego enjuagar bien en agua desionizada y etanol y dejar secar al aire durante al menos 1 h.
- Construimos una simple célula de flujo a la casa de las células en el microscopio. Esto implica el trazado de líneas de grasa de parafina en un portaobjetos de vidrio BK7 microscopio y luego la creación de un túnel-sandwich, colocando uno de los coverlips limpia en la parte superior, y presionando suavemente con un par de pinzas, dando un volumen de celda de flujo de 5.10 l .
- Para observar las células inmovilizadas llenamos el flujo de las células mediante una inyección con un 0,1% w / v solución de poli-L-lisina, y deje que se incuban a temperatura ambiente durante al menos 1 min. Luego enjuagar con 100 medios mínimos l mediante la inyección de los medios de comunicación de un extremo de la celda de flujo y transporte de humedad con papel de seda de la otra.
- A continuación, la mecha a través de 20 l de una dilución 1:500 de 200 nm de diámetro microesferas de látex (Polysciences) en medios mínimos para marcar la superficie cubreobjetos. La celda de flujo se invierte de tal manera que el cubreobjetos se encuentra hacia abajo, colocar en una plataforma clara de la superficie en una cámara sencilla de humedad, y se incuban a temperatura ambiente durante 5 min. Cuentas no consolidadas se eliminan por lavado por absorción a través de 100 medios mínimos l.
- Si queremos observar células atados omitimos el paso de poli-L-lisina y en lugar de llenar la celda de flujo con 5 mg / ml de anticuerpos anti-flagelina. La celda de flujo es un lugar en la cámara de humedad durante 10 minutos y luego se eliminan a través de absorción.
- 20 l de cultivo de células es entonces malo a través de la celda de flujo, ya sea usando la muestra cortada si la observación de las células atados o la muestra unsheared en ver células inmovilizadas.
- La celda de flujo se invierte y se coloca en la cámara de humedad durante 20 minutos. Las células no unidas se lavan a cabo por absorción a través de 100 medios mínimos l.
- Una gota de aceite de inmersión se coloca en el centro de la superficie superior del cubreobjetos y el flujo de las células se coloca suavemente sobre el soporte de muestras de la costumbre microscopio de fluorescencia construido, haciendo contacto con la óptica de lentes de alta apertura numérica objetivo.
- El microscopio electrónico, multiplicando la cámara se enciende y la cámara en que se enfrió a -70 º C, el software está listo para adquirir imágenes a una velocidad típica de 25 Hz en modo de cuadro de transferencia, en un principio disenabling el control de ganancia en el cámara.
- La iluminación de campo claro está encendido y la imagen se pusieron sobre la mesa, la selección de una célula adecuada o de un grupo de células para formar una imagen sobre la base de su ser atrapado con su eje paralelo a la superficie a largo cubreobjetos. El objetivo es ajustar con precisión para asegurar que las 200 cuentas nm látex pegado a la superficie del cubreobjetos son sólo en el enfoque.
- Una secuencia de imágenes se adquieren en el campo claro para grabar el contorno del cuerpo celular. La iluminación de campo claro se apaga y la ganancia de la cámara se activa al máximo.
- Para una adquisición estándar mediante fluorescencia de reflexión total interna (TIRF o) un rayo láser en las longitudes de onda (en este caso, para la excitación de la proteína verde fluorescente, se utiliza un láser nm 473) se pre-establecido con el fin de centrarse en el plano focal posterior de la lente del objetivo, pero desplazados lateralmente desde el eje óptico para generar un campo evanescente de excitación de fluorescencia en la muestra de células.
- La adquisición de la cámara se inicia y se abre el obturador de láser para excitar a las proteínas fluorescentes en las bacterias. Los parámetros de la intensidad del láser y la velocidad de adquisición deben ser optimizados para el sistema biológico particular bajo investigación por experimentar con diferentes valores, pero un típico rango rElevant al estudio de los complejos móviles proteína en la membrana celular son la energía láser 1.10 mW en una zona de excitación circular de diámetro aproximadamente 30 micras, con un tiempo de exposición por cuadro de 50 a 40 ms. Las muestras se iluminan hasta photobleached, por lo general de unos 10 s.
- Este protocolo se puede utilizar tanto para las células atados en la que el cuerpo de la célula está girando alrededor de un punto de unión entre el cubreobjetos y un talón de flagelos, y por células inmovilizadas que están rígidamente fijado a la superficie cubreobjetos.
- Algunas cepas de células que tienen un alto número de copias de los complejos se benefician de un láser blanqueador inicial limitada por difracción se centraron en un polo de la célula antes de la TIRF imágenes. Este blanqueo es equivalente a la utilizada en la recuperación de la fluorescencia después de photobleaching (o FRAP). Utilizando nuestro microscopio costumbre parte de la luz láser de excitación puede ser alimentado de en una segunda vía independiente utilizada para el blanqueo de tipo FRAP. Por lo general 1-10 mW de potencia láser se utiliza, con un tiempo de blanqueo típica en el rango de 10 a 300 ms. Esto se traduce en contraste de imagen mucho mayor en la zona de blanqueado de la célula que permite complejos individuales que posteriormente se difunden en la zona a ser más fácil de visualizar.
- Para visualizar los complejos en el citoplasma de la célula que se difunden mucho más rápido que los de la membrana celular se emplea un modo de iluminación diferente llamado "slimfield". Aquí un láser enfocado es expandido lateralmente para abarcar una sola célula. Esto produce un campo muy intenso que permite la exposición mucho más rápido de lo general una milésima de segundo que deban tomarse.
- Tras la adquisición de datos, las imágenes se introducen en el software personalizado por escrito (codificado en LabVIEW 8.5). Este detecta automáticamente las posiciones de puntos fluorescentes en las células con una precisión de unos pocos nanómetros general y los extractos de su tamaño y brillo. El brightnessof es el trazo photobleaching con respecto al tiempo de un complejo molecular seguimiento para estimar la estequiometría, es decir, la cantidad de proteínas fluorescentes individuales constituyen un complejo molecular única.
Los resultados representativos:
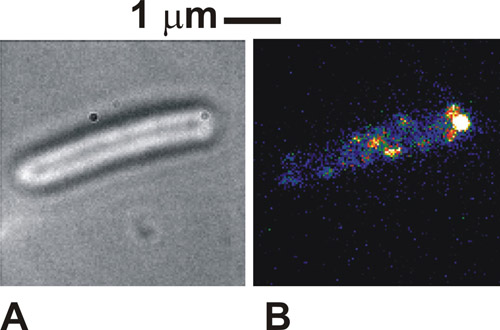
Cuando el protocolo se hace correctamente las imágenes de las células vistas en campo claro es muy distinto, con los perímetros de los cuerpos celulares oscuro contra un cuerpo de la célula blanco / gris (Figura 1a). En la fluorescencia utilizando células inmovilizadas, podemos ver la intensidad de los puntos distintos, por lo general de 250 a 300 nm de ancho (Figura 1b). Las células sanas, sujetos, se verá a girar alrededor del punto de unión de sujeción en las imágenes de campo claro. En virtud de excitación de fluorescencia algunos complejos moleculares, en nuestro caso también podría ser visto en el punto de unión, lo que indica una localización de la proteína marcada con el motor flagelar. Estos puntos son complejos moleculares individuales y el número de visto dependerá del modo de iluminación utilizados y cómo muchos de los complejos están presentes en la célula en un momento dado. La movilidad de los lugares depende del sistema biológico específico en estudio. Si la densidad de puntos es inicialmente muy alto, como es el caso de los citocromos etiqueta se utiliza aquí, a continuación, realizar un blanqueador FRAP inicial puede mejorar el contraste de la imagen.

Figura 1. (A) claro y (B) la imagen TIRF (falso color) de una célula de Escherichia coli inmovilizada expresar una proteína de fusión a la proteína verde fluorescente (GFP), que se sabe están involucrados en los motores de flagelos de las bacterias. Por favor, haga clic aquí para ver una versión ampliada de la figura 1.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Se debe tener cuidado de no "sobre corte" de células para observar las bacterias atados, ya que esto puede perjudicar la funcionalidad de los motores del flagelo. Es importante la utilización de las células durante mucho más tiempo de una hora una vez en el portaobjetos del microscopio, ya que puede convertirse en falta de oxígeno. Optimización considerable puede ser requerido para encontrar las mejores condiciones de microscopio de imagen atiende a su sistema biológico específico bajo investigación. Puede ser aconsejable el intento de la imagen usando purificado GFP sola para determinar la intensidad del láser de excitación necesaria para corregir el sistema de microscopio particular.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Reconocemos los donativos en especie de las cepas bacterianas de los grupos de la Prof. Judith Armitage (Universidad de Oxford, Reino Unido) y el Prof. Conrad Mullineaux (Universidad Queen Mary de Londres, Reino Unido). IMD es financiado conjuntamente por el Departamento de Bioquímica (Universidad de Oxford) y OCISB; AR es financiado por una Ingeniería y Ciencias Físicas Research Council (EPSRC) becas para estudios de DTC, ND se financia con la Biotecnología y Ciencias Biológicas de Investigación (BBSRC), MCL es financiado por una beca Real Sociedad de Investigación de la Universidad.
References
- Leake, M. C., Chandler, J. H., Wadhams, G. H., Bai, F., Berry, R. M., Armitage, J. P. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Leake, M. C., Greene, N. P., Godun, R. M., Granjon, T., Buchanan, G., Chen, S., Berry, R. M., Palmer, T., Berks, B. C. Variable stoichiometry of the TatA component of the twin-arginine protein transport system observed by in vivo single-molecule imaging. Proc Natl Acad Sci U S A. 105, 15376-15381 (2008).
- Leake, M. C., Wilson, D., Gautel, M., Simmons, R. M., M, R. The elasticity of single titin molecules using a two-bead optical tweezers assay. Biophys. J. 87, 1112-1135 (2004).
- Leake, M. C., Wilson, D., Bullard, B., Simmons, R. M. The elasticity of single kettin molecules using a two-bead laser-tweezers assay. FEBS Lett. , 535-555 (2003).
- Lenn, T., Leake, M. C., Mullineaux, C. W. In vivo clustering and dynamics of cytochrome bd complexes in the Escherichia coli plasma membrane. Mol. Microbiol. 70, 1397-1407 (2008).
- Lenn, T., Leake, M. C., Mullineaux, C. W. Are Escherichia coli OXPHOS complexes concentrated in specialised zones within the plasma membrane. Biochem. Soc. Trans. 36, 1032-1036 (2008).
- Lo, C. J., Leake, M. C., Pilizota, T., Berry, R. M. Single-cell measurements of Membrane Potential, Sodium-Motive Force and Flagellar Motor Speed in Escherichia coli. Biophys. 93, 294-302 (2007).
- Lo, C. J., Leake, M. C., Berry, R. M. Fluorescence measurement of intracellular sodium concentration in single Escherichia coli cells. Biophys. J. 90, 357-3565 (2006).

{kind=link}