

Summary
Ici nous démontrons les protocoles pour effectuer une seule molécule de microscopie par fluorescence sur des cellules vivantes pour permettre aux bactéries fonctionnelles complexes moléculaires à détecter, suivre et quantifiés.
Abstract
Aperçu complet sur les mécanismes de cellules vivantes peuvent être atteints que par l'étude des processus clés qui déclenchent et direct d'événements au niveau cellulaire. À ce jour, la complexité des systèmes biologiques de cisaillement a provoqué précise molécule unique d'expérimentation à être beaucoup trop exigeant, de se concentrer plutôt sur des études de systèmes simples à l'aide en vrac relativement rudimentaires ensemble de la moyenne des mesures. Toutefois, plusieurs processus importants se produisent dans la cellule vivante, au niveau d'un seul ou de quelques molécules; mesures d'ensemble masque généralement la nature stochastique et hétérogène de ces événements. Ici, en utilisant la microscopie optique de pointe et d'analyse des outils d'analyse d'image, nous démontrons comment surveiller les protéines au sein d'une seule cellule vivante bactérienne avec une précision de molécules uniques et comment nous pouvons observer la dynamique au sein de complexes moléculaires dans le fonctionnement des machines biologiques. Les techniques sont directement pertinents physiologiquement. Ils sont peu-perturbatifs et non-invasive à l'échantillon biologique à l'étude et sont entièrement à l'écoute pour des recherches dans la matière vivante, les fonctionnalités ne sont pas facilement disponibles pour les autres une seule molécule des approches de biophysique. En outre, les échantillons biologiques étudiés tous les produits de protéines par fluorescence marqués à des niveaux qui sont presque identiques aux souches de cellules non modifiées («codage» génomique), par opposition à l'approche la plus commune, mais moins idéal pour générer des protéines beaucoup plus que ne se produisent naturellement («plasmide d'expression»). Ainsi, les échantillons biologiques réelles qui seront étudiés sont beaucoup plus près les organismes naturels, et donc la plus pertinente pour les observations réelles des processus physiologiques.
Protocol
- Pour commencer cette procédure, 50 pl de stocks congelés de la protéine fluorescente Escherichia coli exprimant les cellules bactériennes sont d'abord décongelée et culture aérobie avec agitation dans 5 ml de milieu LB de croissance nuit à 37 ° C. Dans la matinée, 50 pi de cette culture saturée est extrait et sous-culture dans les milieux de culture minimale M63 glucose, l'incubation à 30 degrés C pendant 4 à 6 heures. Ici nous démontrons l'aide de deux souches de cellules différentes, dont l'un exprime un transport d'électrons du cytochrome fusionnée à la GFP, l'autre qui exprime une protéine impliquée dans le moteur flagellaire des bactéries fusionnée à la GFP.
- Les cellules peuvent soit récoltée directement de la sous-culture de plus en plus, si les considérant comme des échantillons immobilisés, ou ils peuvent être cisaillées pour tronquer flagelles bactériens Si la visualisation sous la rubrique «captif» des conditions.
- Cisaillement consiste à placer généralement 1-5ml de la sous-culture dans un dispositif constitué de deux seringues stériles rejoint par une tuyauterie stérile. La tonte se fait en alternance dans chacun poussant pompe seringue pour pousser la culture à travers le tuyau étroit. Ceci est fait de 50 à 100 fois, selon l'étendue de cisaillement requise. La culture est ensuite centrifugée pour sédimenter les cellules, qui est remis en suspension dans un milieu minimal pour éliminer les fragments de flagelles.
- Nous préparons ensuite nettoyé BK7 lamelles de verre en la plongeant dans une solution saturée de KOH dans l'éthanol pendant 20 min, puis rincer abondamment à l'eau désionisée et de l'éthanol et laisser sécher à l'air pendant au moins 1 h.
- Nous construisons un simple flux de cellules à la maison des cellules au microscope. Cela implique de tracer des lignes de graisse de paraffine sur une lame de verre BK7 microscope et en créant ensuite un tunnel-sandwich en plaçant l'un des coverlips nettoyé sur le dessus, et en appuyant doucement avec une paire de pinces, ce qui donne un volume cellulaire débit de 50 à 10 ul .
- Pour observer des cellules immobilisées, nous remplissons le flux de cellules par injection avec un 0,1% p / v solution de poly-L-lysine, et lui permettre d'incuber à température ambiante pendant au moins 1 min. Nous avons ensuite rincer à l'aide de 100 ul milieu minimal en injectant les médias d'un bout de la circulation des cellules et de mèche avec du papier absorbant de l'autre.
- Nous avons ensuite la mèche à 20 pi d'une dilution 1:500 de 200 nm de diamètre des microsphères de latex (Polysciences) dans un milieu minimal pour marquer la surface lamelle. Le flux de cellules est inversée de telle sorte que la lamelle est orienté vers le bas, placé sur une plate-forme claire de la surface dans une chambre humide simple, et incubée à température ambiante pendant 5 min. Unbound perles sont ensuite emportées par capillarité à travers les médias 100 ul minime.
- Si nous voulons observer des cellules captif nous omettons l'étape de poly-L-lysine et au lieu de remplir le flux des cellules avec 5 mg / ml d'anticorps anti-flagelline. Le flux de cellules est placé dans la chambre humide pendant 10 min et est ensuite rincé à travers une mèche.
- 20 ul de la culture cellulaire est alors méchants à travers le flux de cellules, soit en utilisant l'échantillon cisaillé, si l'observation des cellules à l'attache ou l'échantillon cisaillé dans la visualisation des cellules immobilisées.
- Le flux de cellules est inversé et placé dans la chambre humide pendant 20 min. Unbound cellules sont ensuite lavées par capillarité à travers les médias 100 ul minime.
- Une goutte d'huile d'immersion est placé dans le centre de la surface supérieure de la lamelle et le flux de cellules est ensuite placé délicatement sur le porte-échantillon du microscope à fluorescence intégré personnalisée, la prise de contact optique avec la lentille de grande ouverture numérique objectif.
- Le microscope électronique multipliant appareil est allumé et l'appareil a été réglée pour être refroidi à -70 ° C, le logiciel est mis à acquérir des images à une cadence typique de 25 Hz en mode de transfert cadre, initialement disenabling le contrôle de gain sur la caméra.
- L'éclairage fond clair est allumé et l'image est mis en évidence, en sélectionnant une cellule appropriée ou d'un groupe de cellules à imager sur la base de leur être coincés avec leur axe parallèle à la surface à long lamelle. L'accent est finement ajusté afin de s'assurer que les 200 billes de latex nm collé à la surface lamelle sont juste à point.
- Une séquence d'image est acquise en fond clair d'enregistrer le contour du corps cellulaire. L'éclairage fond clair est éteint et le gain de la caméra est activé au maximum.
- Pour une acquisition standard à l'aide totale à fluorescence à réflexion interne (ou la FRBR) un faisceau laser sur longueur d'onde appropriée (ici, pour l'excitation protéine fluorescente verte, nous utilisons un laser nm 473) est pré-établis de manière à se concentrer dans le plan focal arrière de la l'objectif, mais déplacées latéralement de l'axe optique pour générer un champ évanescent d'excitation de fluorescence dans l'échantillon de cellules.
- L'acquisition de la caméra est démarré et l'obturateur ouvert au laser pour exciter les protéines fluorescentes dans les bactéries. Les paramètres de l'intensité du laser et la vitesse d'acquisition doivent être optimisés pour le système biologique particulier sous enquête par l'expérimentation avec des valeurs différentes, mais une gamme typique rPERTINENTES à l'étude de complexes protéiques mobiles dans la membrane cellulaire sont de 10 à 10 mW de puissance laser sur une zone d'excitation circulaire de diamètre ~ 30 um, avec un temps d'exposition par image de 5-40 ms. Les échantillons sont éclairés jusqu'à ce photobleached, généralement pour ~ 10 s.
- Ce protocole peut être utilisé aussi bien pour les cellules captif dans lequel le corps de la cellule est en rotation autour d'un point d'attache entre la lamelle et un talon flagellaire, et pour des cellules immobilisées qui sont rigidement fixées à la surface lamelle.
- Certaines souches de cellules qui ont un nombre élevé de copies de complexes de bénéficier d'une eau de Javel initiale limitée par la diffraction laser focalisé sur un pôle de la cellule avant de la FRBR imagerie. Cette eau de Javel est équivalent à celui utilisé dans la récupération de fluorescence après photoblanchiment (ou PAF). En utilisant notre microscope personnalisée partie de la lumière laser d'excitation peut être alimenté au loin dans un second chemin indépendant utilisé pour FRAP type de blanchiment. Typiquement 1-10mW de puissance laser est utilisé, avec un temps de javel typique dans la gamme 10-300 ms. Ce résultat contraste d'imagerie beaucoup plus élevé dans la zone blanchi de la cellule permettant complexes individuels qui par la suite diffuser dans ce domaine pour être plus facilement visible.
- Pour visualiser les complexes dans le cytoplasme de la cellule qui diffusent beaucoup plus vite que ceux de la membrane cellulaire un mode d'éclairage différent appelé "slimfield" est employé. Voici un laser focalisé est élargi latéralement pour simplement couvrir une seule cellule. Cela produit un champ très intense permettant des expositions beaucoup plus rapide d'une milliseconde généralement à prendre.
- Suite à l'acquisition de données, les images sont introduites dans un logiciel créé sur mesure (code dans LabVIEW 8.5). Il détecte automatiquement les positions des taches fluorescentes dans les cellules avec une précision de quelques nanomètres et quelques extraits généralement leur taille et leur luminosité. Le brightnessof la trace photoblanchiment par rapport au temps d'un complexe moléculaire est suivi ensuite utilisée pour estimer la stoechiométrie, à savoir combien individuels protéines fluorescentes forment un complexe moléculaire unique.
Les résultats représentatifs:
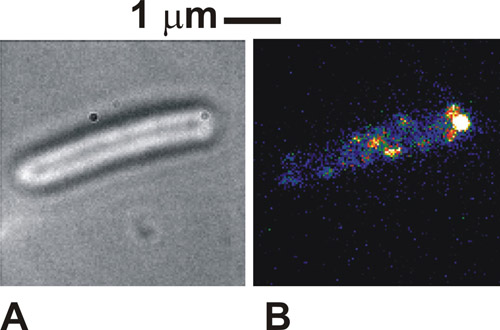
Lorsque le protocole est fait correctement les images des cellules considérées en fond clair est très distincte, avec les périmètres des organismes de cellule sombre contre un corps cellulaire blanc / gris (figure 1a). En utilisant la fluorescence des cellules immobilisées, nous pouvons voir l'intensité des taches distinctes, généralement d'une largeur de 250-300 nm (figure 1b). Sains, les cellules captif sera vu à tourner autour du point d'attache d'ancrage dans les images en fond clair. Sous l'excitation de fluorescence des complexes moléculaires dans notre cas pourrait aussi être vu au point de fixation, indiquant une localisation de la protéine marqués avec le moteur flagellaire. Ces taches sont individuelles complexes moléculaires et le nombre d'entre eux vu dépendra du mode d'éclairage utilisé et combien de complexes sont effectivement présents dans la cellule à un moment donné. La mobilité des taches dépend du système biologique spécifique à l'étude. Si la densité de points est initialement très élevé, comme c'est le cas avec les cytochromes marqué utilisé ici, puis en effectuant une eau de Javel FRAP initiale peut améliorer le contraste d'imagerie.

Figure 1. (A) clair et (B) l'image TIRF (fausses couleurs) pour une cellule d'Escherichia coli immobilisé exprimant une protéine fusionnée à la protéine fluorescente verte (GFP) qui est connue pour être impliquée dans les moteurs flagellaires de bactéries. S'il vous plaît cliquez ici pour voir une version agrandie de la figure 1.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Des précautions doivent être prises pour ne pas «plus de cisaillement" cellule pour regarder les bactéries attachées, car cela pourrait nuire à la fonctionnalité des moteurs flagellaires. Il est important d'utiliser des cellules pour beaucoup plus d'une heure une fois sur la lame du microscope, car ils peuvent devenir pauvre en oxygène. Optimisation considérables peuvent être nécessaires pour trouver les meilleurs conditions d'imagerie microscope traiteur à votre système biologique spécifique de l'enquête. Il peut être sage de tenter l'imagerie utilisant purifiée GFP seule de déterminer l'intensité de l'excitation correcte au laser requise pour votre système de microscope particulier.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Nous reconnaissons les dons en nature des souches bactériennes à partir des groupes du professeur Judith Armitage (Université d'Oxford, Royaume-Uni) et le professeur Conrad Mullineaux (Queen Mary University of London, UK). IMD est financé conjointement par le Département de biochimie (Université d'Oxford) et OCISB; AR est financé par une ingénierie et sciences physiques Research Council (EPSRC) stagiaire de DTC; ND est financé à partir des biotechnologies et des sciences biologiques du Conseil de recherches (BBSRC); MCL est financée par une bourse de la Société royale de recherche universitaire.
References
- Leake, M. C., Chandler, J. H., Wadhams, G. H., Bai, F., Berry, R. M., Armitage, J. P. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Leake, M. C., Greene, N. P., Godun, R. M., Granjon, T., Buchanan, G., Chen, S., Berry, R. M., Palmer, T., Berks, B. C. Variable stoichiometry of the TatA component of the twin-arginine protein transport system observed by in vivo single-molecule imaging. Proc Natl Acad Sci U S A. 105, 15376-15381 (2008).
- Leake, M. C., Wilson, D., Gautel, M., Simmons, R. M., M, R. The elasticity of single titin molecules using a two-bead optical tweezers assay. Biophys. J. 87, 1112-1135 (2004).
- Leake, M. C., Wilson, D., Bullard, B., Simmons, R. M. The elasticity of single kettin molecules using a two-bead laser-tweezers assay. FEBS Lett. , 535-555 (2003).
- Lenn, T., Leake, M. C., Mullineaux, C. W. In vivo clustering and dynamics of cytochrome bd complexes in the Escherichia coli plasma membrane. Mol. Microbiol. 70, 1397-1407 (2008).
- Lenn, T., Leake, M. C., Mullineaux, C. W. Are Escherichia coli OXPHOS complexes concentrated in specialised zones within the plasma membrane. Biochem. Soc. Trans. 36, 1032-1036 (2008).
- Lo, C. J., Leake, M. C., Pilizota, T., Berry, R. M. Single-cell measurements of Membrane Potential, Sodium-Motive Force and Flagellar Motor Speed in Escherichia coli. Biophys. 93, 294-302 (2007).
- Lo, C. J., Leake, M. C., Berry, R. M. Fluorescence measurement of intracellular sodium concentration in single Escherichia coli cells. Biophys. J. 90, 357-3565 (2006).

{kind=link}