

Summary
Aqui demonstramos os protocolos para a realização de uma única molécula de microscopia de fluorescência em células vivas de bactérias para habilitar funcionais complexos moleculares a serem detectados, rastreados e quantificados.
Abstract
Pleno conhecimento sobre os mecanismos de células vivas só pode ser alcançada por investigar os processos-chave que provocam direta e eventos em um nível celular. Até o momento a complexidade de cisalhamento de sistemas biológicos tem causado experimentação molécula única precisa ser demasiado exigentes, em vez enfocando estudos de sistemas simples utilizando granel relativamente rudimentar ensemble da média das medições. No entanto, muitos processos importantes ocorrem na célula viva no nível de apenas uma ou algumas poucas moléculas; medições ensemble geralmente mascaram a natureza estocástica e heterogénea desses eventos. Aqui, utilizando microscopia óptica avançada e ferramentas de análise de análise de imagens que demonstram como monitorar proteínas dentro de uma única célula viva bacteriana com uma precisão de moléculas individuais e como podemos observar a dinâmica dentro de complexos moleculares em funcionamento máquinas biológicas. As técnicas são diretamente relevantes fisiologicamente. Eles são minimamente perturbativos e não-invasivo para a amostra biológica em estudo e estão totalmente sintonizadas para as investigações na vida material, recursos não disponíveis para outros uma única molécula de abordagens de biofísica. Além disso, as amostras biológicas estudadas todas as produzem a proteína fluorescente marcadas em níveis que são quase idênticas às cepas de células não modificadas ("codificação genômica"), ao contrário da abordagem mais comum, mas menos ideal para a geração de proteínas significativamente mais do que ocorreria naturalmente ('expressão plasmídeo). Assim, as amostras biológicas reais que serão investigadas são significativamente mais dos organismos naturais, e, portanto, as observações mais relevantes para os reais processos fisiológicos.
Protocol
- Para iniciar este procedimento, 50 ul de estoques congelados de proteína fluorescente expressar Escherichia coli células bacterianas são os primeiros descongelados e cresceu aerobicamente com agitação em 5 ml de mídia LB crescimento durante a noite a 37 graus C. Pela manhã, 50 ul desta cultura saturada é extraído e sub-cultivadas em um mínimo M63 media glucose cultura, incubando a 30 graus C para 4 a 6 horas. Aqui demonstramos usando duas linhagens celulares diferentes, um dos quais exprime um transporte de elétrons do citocromo fundido a GFP, o outro que expressa uma proteína envolvida no motor flagelar bacteriano fundido a GFP.
- Células podem ou colhidas diretamente do crescente sub-cultura se vê-las como amostras de imobilizado, ou podem ser cortados para truncar flagelos bacterianos se ver em "amarrados" condições.
- Shearing tipicamente envolve a colocação de 1-5ml da sub-cultura em um dispositivo composto por duas seringas estéreis unidas por tubos estéreis. O corte é feito pela alternância empurrando em cada bomba de seringa para empurrar a cultura através do tubo estreito. Isto é feito 50-100 vezes, dependendo da extensão do corte necessário. A cultura é então centrifugado para agregar as células, que é ressuspenso em meio mínimo para remover fragmentos de flagelos.
- Nós, então, preparar limpos BK7 lamínulas de vidro por imersão em uma solução saturada de KOH em etanol por 20 min, em seguida, enxaguar bem em água desionizada e etanol e deixar secar ao ar por pelo menos 1 h.
- Nós construímos uma simples célula de fluxo para a casa das células ao microscópio. Isto envolve o desenho de linhas de graxa de parafina em uma lâmina de microscópio BK7 vidro e em seguida, criar um túnel sanduíche colocando uma das coverlips limpo em cima e pressionando delicadamente com uma pinça, dando um volume de célula de fluxo de 50-10 mL .
- Para observar células imobilizadas enchemos o fluxo de células por injeção com uma solução de 0,1% w / v de poli-L-lisina, e permitir que ele deixe em temperatura ambiente por pelo menos 1 min. Em seguida, lave com o uso mínimo de 100 l de mídia, injetando os meios de comunicação de uma extremidade da célula de fluxo e absorção com papel de tecido da outra.
- Em seguida, através de pavio 20 l de uma diluição de 1:500 de 200 nm de diâmetro microesferas de látex (Polysciences) em meio mínimo para marcar a superfície lamela. A célula de fluxo é invertida de modo que a lamela está voltada para baixo, colocado sobre uma plataforma clara da superfície em uma câmara de umidade simples, e incubadas em temperatura ambiente por 5 min. Contas não ligados são então lavados por absorção através de 100 l media mínima.
- Se quisermos observar as células amarrados omitimos o passo poli-L-lisina e, em vez encher a célula de fluxo com 5 mg / ml de anticorpos anti-flagelina. A célula de fluxo é lugar na câmara úmida por 10 min e depois é lavado por meio de absorção.
- 20 l da cultura de células é então mau através da célula de fluxo, utilizando a amostra cisalhada se observar células de presos ou a amostra unsheared em ver células imobilizadas.
- A célula de fluxo é invertido e colocado na câmara úmida por 20 min. Células não ligados são então lavados por absorção através de 100 l media mínima.
- Uma gota de óleo de imersão é colocada no centro da superfície superior da lamela eo fluxo de células é então colocada gentilmente sobre o porta-amostras do microscópio construído sob encomenda de fluorescência, fazendo contato óptico com a objectiva de alta abertura numérica.
- O microscópio de elétron-multiplicando câmera é ligada e definida para a câmera ser resfriado a -70 graus C, o software está definido para adquirir imagens a uma taxa de quadros típicos de 25 Hz no quadro de transferência de modo, inicialmente disenabling o controle de ganho na câmera.
- A iluminação de campo claro é ligado ea imagem é colocada em foco, selecionando uma célula adequada ou grupo de células a ser trabalhada com base em seu ser preso com o seu longo eixo paralelo à superfície da lamínula. O foco é ajustado com precisão para garantir que as 200 contas nm latex preso à superfície lamela são apenas em foco.
- Uma seqüência de imagens é adquirido em campo claro para gravar o contorno do corpo celular. A iluminação de campo claro está desligado eo ganho câmera é ativada ao máximo.
- Para uma aquisição padrão usando fluorescência de reflexão interna total (ou TIRF) um feixe de laser sobre comprimento de onda apropriado (aqui, para a excitação da proteína verde fluorescente, usamos um laser de 473 nm) é pré-fixado de modo a ser focado no plano focal posterior da a lente objetiva, mas deslocadas lateralmente do eixo óptico para gerar um campo evanescente de excitação de fluorescência na amostra de células.
- A aquisição da câmera é iniciado eo obturador aberto a laser para excitar as proteínas fluorescentes no interior da bactéria. Os parâmetros para a intensidade do laser ea velocidade de aquisição precisa ser otimizado para o sistema particular sob investigação biológica através da experimentação de diferentes valores, mas uma gama típica relevant a estudar complexos de proteína celular na membrana celular são a potência do laser 10-10 mW sobre uma área circular de excitação de diâmetro ~ 30 mm, com um tempo de exposição por quadro de 50-40 ms. Amostras são iluminadas até Foto-descoloração, tipicamente para ~ 10 s.
- Este protocolo pode ser utilizado tanto para as células amarrados em que o corpo da célula está girando sobre um ponto de ligação entre as lamelas e um esboço flagelar, e por células imobilizadas que são rigidamente fixados à superfície lamela.
- Algumas cepas de células que têm um número elevado de cópias de complexos beneficiar de uma lixívia de laser inicial de difração limitada focada em um pólo da célula antes TIRF imagem. Esta lixívia é equivalente ao utilizado na recuperação de fluorescência após a fotodegradação (ou FRAP). Usando nosso microscópio personalizado um pouco da luz do laser de excitação pode ser alimentado fora em um segundo caminho independente usado para FRAP tipo de clareamento. Normalmente 1-10mW de potência do laser é usado, com um tempo de lixívia típico na faixa de 10-300 ms. Isso resulta em contraste de imagem muito maior na zona branqueada da célula permitindo complexos individuais, que posteriormente difusa em que a área a ser mais facilmente visualizadas.
- Para visualizar complexos no citoplasma da célula, que difundem muito mais rápido do que os da membrana da célula um modo de iluminação diferente chamado "slimfield" é empregado. Aqui, um laser focalizado é expandido lateralmente para apenas abranger uma única célula. Isso produz um campo muito intenso permitindo exposições muito mais rápido do normalmente um milésimo de segundo a tomar.
- Após a aquisição de dados, as imagens são alimentados em software escrito personalizado (codificados em LabVIEW 8.5). Este detecta automaticamente as posições dos pontos fluorescentes nas células para uma precisão de um nanômetros geralmente poucos e extratos de seu tamanho e brilho. O brightnessof o traço fotobranqueamento com respeito ao tempo de um complexo molecular rastreado é então usada para estimar a estequiometria, ou seja, quantos individuais proteínas fluorescentes compõem um único complexo molecular.
Resultados representativos:
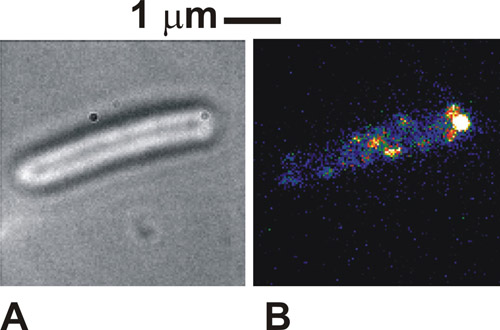
Quando o protocolo é feito corretamente as imagens das células vista em campo claro é muito distintos, com os perímetros dos corpos cela escura contra um corpo celular branco / cinza (Figura 1a). Na fluorescência usando células imobilizadas, podemos ver a intensidade pontos distintos, tipicamente de 250-300 nm de largura (Figura 1b). Saudável, as células amarrados será visto para girar em torno do ponto de fixação tether em imagens de campo claro. Sob excitação de fluorescência alguns complexos moleculares no nosso caso também pode ser visto no ponto de ligação, indicando uma localização da proteína marcada com o motor flagelar. Essas manchas são individuais complexos moleculares e do número deles visto vai depender do modo de iluminação usados e como muitos dos complexos estão realmente presentes na célula de cada vez. A mobilidade dos pontos depende do sistema biológico específico em estudo. Se a densidade de pontos é inicialmente muito alta, como é o caso com os citocromos rotulados usado aqui, então executar uma lixívia FRAP inicial pode melhorar o contraste de imagem.

Figura 1. (A) Brightfield e (B) imagem TIRF (cor falsa) de uma célula imobilizada Escherichia coli expressando uma proteína fundida a proteína verde fluorescente (GFP), que é conhecida por estar envolvida nos motores flagelares de bactérias. Por favor, clique aqui para ver uma versão ampliada da figura 1.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
É preciso ter cuidado para não "sobre cisalhamento" célula para olhar as bactérias amarrados, pois isso pode prejudicar a funcionalidade dos motores flagelares. É importante o uso de células por muito mais tempo do que uma hora uma vez na lâmina de microscópio, uma vez que pode tornar-se falta de oxigênio. Otimização considerável pode ser necessária para encontrar as melhores condições de imagem do microscópio servidos ao seu sistema biológico específico sob investigação. Pode ser sábio para tentar a imagem usando GFP purificada sozinho para verificar a excitação do laser correta intensidade necessária para o seu sistema de microscópio particular.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Reconhecemos as doações tipo de cepas de bactérias do grupo do Prof Judith Armitage (Universidade de Oxford, UK) e Conrad Prof Mullineaux (Queen Mary University of London, UK). IMD é financiado conjuntamente pelo Departamento de Bioquímica (Universidade de Oxford) e OCISB; AR é financiado por uma Engenharia e Ciências Físicas Research Council (EPSRC) studentship DTC; ND é financiado pelo Biotecnologia e Ciências Biológicas Research Council (BBSRC); MCL é financiada por uma Universidade Royal Society Fellowship Research.
References
- Leake, M. C., Chandler, J. H., Wadhams, G. H., Bai, F., Berry, R. M., Armitage, J. P. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 443, 355-358 (2006).
- Leake, M. C., Greene, N. P., Godun, R. M., Granjon, T., Buchanan, G., Chen, S., Berry, R. M., Palmer, T., Berks, B. C. Variable stoichiometry of the TatA component of the twin-arginine protein transport system observed by in vivo single-molecule imaging. Proc Natl Acad Sci U S A. 105, 15376-15381 (2008).
- Leake, M. C., Wilson, D., Gautel, M., Simmons, R. M., M, R. The elasticity of single titin molecules using a two-bead optical tweezers assay. Biophys. J. 87, 1112-1135 (2004).
- Leake, M. C., Wilson, D., Bullard, B., Simmons, R. M. The elasticity of single kettin molecules using a two-bead laser-tweezers assay. FEBS Lett. , 535-555 (2003).
- Lenn, T., Leake, M. C., Mullineaux, C. W. In vivo clustering and dynamics of cytochrome bd complexes in the Escherichia coli plasma membrane. Mol. Microbiol. 70, 1397-1407 (2008).
- Lenn, T., Leake, M. C., Mullineaux, C. W. Are Escherichia coli OXPHOS complexes concentrated in specialised zones within the plasma membrane. Biochem. Soc. Trans. 36, 1032-1036 (2008).
- Lo, C. J., Leake, M. C., Pilizota, T., Berry, R. M. Single-cell measurements of Membrane Potential, Sodium-Motive Force and Flagellar Motor Speed in Escherichia coli. Biophys. 93, 294-302 (2007).
- Lo, C. J., Leake, M. C., Berry, R. M. Fluorescence measurement of intracellular sodium concentration in single Escherichia coli cells. Biophys. J. 90, 357-3565 (2006).

{kind=link}