

Summary
Intra-tumoral heterogenitet er et iboende træk ved tumorer, herunder gliomer. Vi udviklede en enkel og effektiv protokol, der bruger en kombination af buffere og gradient centrifugering til at isolere enkelt kerner fra friske frosne gliomvæv til enkelt kerne RNA og ATAC sekventering undersøgelser.
Abstract
Voksne diffuse gliomer udviser inter- og intra-tumor heterogenitet. Indtil for nylig har størstedelen af storstilet molekylær profileringsindsats fokuseret på bulktilgange, der førte til molekylær klassificering af hjernetumorer. I løbet af de sidste fem år, enkelt celle sekventering tilgange har fremhævet flere vigtige funktioner i gliomer. De fleste af disse undersøgelser har udnyttet friske hjernetumorprøver til at isolere enkeltceller ved hjælp af flowcytometri eller antistofbaserede separationsmetoder. Fremadrettet vil brugen af friskfrosne vævsprøver fra biobanker give større fleksibilitet til enkeltcelleapplikationer. Desuden, som encellet felt fremskridt, den næste udfordring vil være at generere multi-omics data fra enten en enkelt celle eller den samme prøve forberedelse til bedre optrævle tumor kompleksitet. Derfor vil enkle og fleksible protokoller, der tillader datagenerering til forskellige metoder såsom single-nucleus RNA sekventering (snRNA-seq) og enkelt kerne Assay for Transposase-Accessible Chromatin med høj-throughput sekventering (snATAC-seq) være vigtigt for feltet.
De seneste fremskridt inden for enkeltcelleområdet kombineret med tilgængelige mikrofluidiske instrumenter såsom 10x genomforskningsplatformen har lettet enkeltcelleapplikationer i forskningslaboratorier. For at studere hjernesvulst heterogenitet, vi udviklet en forbedret protokol for isolering af enkelt kerner fra friske frosne gliomer. Denne protokol fletter eksisterende enkeltcelleprotokoller og kombinerer et homogeniseringstrin efterfulgt af filtrering og buffermedieret gradientcentrifugering. De resulterende prøver er rene enkeltkerneaffjedring, der kan bruges til at generere enkeltkernegenekspression og kromatinadgangsdata fra samme kernepræparat.
Introduction
Diffuse lavere kvalitet gliomer (LGG), den mest almindelige primære hjernesvulst hos voksne, er infiltrerede neoplasmer, der ofte opstår på hjernehalvdelen. LGGs udviser både inter- og intra-tumor heterogenitet, som ikke kun drives af tumorpopulationen, men også af de ikke-maligne celler, der er indviklet involveret i tumorudvikling og progression1,2,3,4,5.
I løbet af det sidste årti har der været en lavine af genomiske data indsamlet inden for gliomer. Disse data stammer hovedsageligt fra bulk tumor sekventering undersøgelser og har bidraget enormt til den molekylære karakterisering, og den nuværende klassificering af hjernetumorer5,6,7,8,9,10,11. Men selv om disse undersøgelser afslørede det brede molekylære landskab forbundet med gliomer, er der stadig en skuffende mangel på fremskridt med hensyn til terapeutisk intervention. En af hindringerne for behandling resistens i hjernen tumorer er intra-tumor heterogenitet. For at løse dette problem har forskellige undersøgelser fokuseret på den genomiske, transskriptomiske, proteomiske og epigenetiske heterogenitet, der er til stede i en tumor på et enkelt celleniveau12,13,14,15,16,17.
Selv om der har været bemærkelsesværdige teknologiske fremskridt i enkeltcelleområdet i løbet af de sidste par år, er en af de vigtigste begrænsende faktorer tilgængeligheden af friske prøver, der er nødvendige for at isolere cellerne og udføre disse eksperimenter. For at overvinde denne begrænsning har der været flere vellykkede forsøg på at udføre analyser, såsom snRNA-seq og snATAC-seq fra frosne væv, ved hjælp af kerner i stedet for cellerne18,19. De fleste af disse metoder er afhængige af enten fluorescensaktiveret cellesortering (FACS) eller filtreringsstrategier. Både enkelt celle og enkelt kerner tilgange har deres styrker og ulemper. Enkelt celle tilgange opretholde mitokondrie udskrifter, som selv kan være informativ, kan også reducere transcriptome dækning på grund af deres høje overflod. Enkelt kernei isolation tilgange fjerne en høj procentdel af mitokondrie fraktion, hvilket giver en mere dybdegående dækning af de nukleare udskrifter20.
Der er forskellige kommercielt tilgængelige platforme, der er blevet brugt i de seneste år til at analysere enkeltcellegenomdata, herunder RNA-seq og ATAC-seq. En af de mest fremtrædende platforme er 10x Genomics Chromium platform for enkeltcellet genekspression og enkeltcellet ATAC-profilering. Da platformen arbejder ved hjælp af mikrofluidiske kamre, kan snavs eller aggregater tilstoppe systemet, der fører til tab af data, reagenser og værdifulde kliniske prøver. Derfor afhænger succesen af enkeltcelleundersøgelser i høj grad af den nøjagtige isolering af enkeltceller / kerner.
Den protokol, som vi vil demonstrere heri, er en lidt modificeret kombination af DroNc-seq- og Omni-ATAC-seq-protokollerne og bruger en lignende tilgang til nylige undersøgelser, der bruger snRNA-seq til at forstå neurologiske lidelser og neuronale celletyper i den menneskelige hjerne18,19,21,22,23,24. Protokollen bruger en kombination af enzymatisk/mekanisk dissociation af frosne prøver efterfulgt af filtrering og gradient centrifugering og giver mulighed for hurtig og præcis isolering af enkelt kerner fra friske frosne gliomvæv. Vi har med succes brugt denne protokol til at generere snRNA-seq og snATAC-seq data fra samme kerner forberedelse fra hjernetumor prøver.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Friske frosne glioomprøver blev fremstillet fra National Center for Tumor Diseases (NCT)-vævsbank i Heidelberg, Tyskland. Brugen af patientmateriale blev godkendt af Institutionsgennemgangsnævnet på Det Medicinske Fakultet i Heidelberg, og der blev indhentet informeret samtykke fra alle patienter, der indgik i undersøgelsen.
1. Eksperimentel forberedelse
- Udfør alle trin på is eller ved 4 °C.
- Prækølingsrør, tallerkener, barberblade, douncers og stødere til 4 °C.
- Forbered alle buffere på forhånd. Disse buffere er stabile ved stuetemperatur. Steril filtrering anbefales, især til saccharose. Lagerforberedelserne ændres fra Corces et al.19. Se tabel 1-7.
- Prøver udtages fra flydende nitrogen eller -80 °C-fryseopbevaring, og der opbevares tøris indtil trin 2.1.
2. Væv dissektion og dissociation
- Overfør frisk frossen vævsprøve (10-60 mg) til en forkølet petriskål. Hakkekød /hak frisk frosset væv med et barberblad til små stykker på is.
- Der tilsættes 500 μL kølet kerner buffer til et forkølet 1,5 mL rør. Vævsstykkerne placeres i 1,5 mL-røret, der indeholder kernelysbufferen, og overføres til en Douncer (Materialetabel).
BEMÆRK: Der er to Dounce homogenisator-stødere: "løs" eller "A"-støder til indledende prøvereduktion og "stram" eller "B"-støder for fuldstændig prøve homogenisering. - Dounce vævsstykkerne med den "løse" støder i ca. 20 slag, indtil friktionen er reduceret. Hvis der er snavs til stede, kan prøven forklares ved filtrering med en 100 mm si.
- Dounce med den "stramme" støder for 20 slag for at opnå fuldstændig væv homogenisering.
- Homogenaten (ca. 500 μL) overføres til et forkølet 2 mL rør. Tilsæt 1 mL kølet lysis buffer. Bland forsigtigt og inkuber på is i 5 min. Bland forsigtigt med en bredboret pipettespids og gentag 1-2 gange under inkubationen.
- Filtrer hele homogenatet ved hjælp af et 30 μm sinet, saml dig i et 15 mL Falcon rør og overfør tilbage til et nyt forkølet 2 mL rør. En enkelt si er typisk tilstrækkelig til hele homogenatet.
- Kontroller under et let mikroskop for at kontrollere fjernelsen af store snavs og den nukleare membrans intakthed. Kerner skal være runde, og atommembranen må ikke forvrænges. Hvis der er snavs til stede, kan kerner filtreres igen.
- Centrifugere kernerne ved 500 x g i 5 min ved 4 °C på en bænk øverste centrifuge. Fjern supernatanten og efterlad ~50 μL med pellet, der indeholder kernerne. Forsigtigt resuspend pellet i en anden 1 mL kerner buffer og inkubere i 5 minutter på is.
- Centrifugere kernerne ved 500 x g i 5 min ved 4 °C. Fjern supernatanten uden forstyrrende pellet, tilsæt 500 mL 1x homogeniseringsbuffer (HB) (tabel 4) og inkuber i 5 min uden genoplivning. Derefter genbruges kernerne i en anden 1,0 mL af 1x HB.
- Centrifugere kernerne ved 500 x g i 5 min ved 4 °C. Supernatanten fjernes, og nuklejerne tages forsigtigt ud af kernerne igen i 200 μL 1x HB i et nyt 2,0 mL rør.
3. Gradient centrifugering
- Der tilsættes 200 μL 50% joxanolopløsning (tabel 5) for at give en endelig koncentration på 25% joxanol. Bland godt 10 gange med pipette indstillet på 300 mL.
- Der tilsættes 300 μL 29% joxanolopløsning (tabel 6) under blandingen på 25 %. Brug en P1000 fin spids for at undgå blanding af lagene.
- Der tilsættes 300 μl 35% joodixanolopløsning (tabel 7) under blandingen på 29 %. Brug en P1000 fin spids for at undgå blanding af lagene.
Forsigtig: Dette trin kræver gradvis fjernelse af pipettespidsen under fugtning for at undgå forskydning af for store mængder. - Sæt prøverne i en svingende skovlcentrifuge, drej i 20 min ved 3.500 x g ved 4 °C med bremsen slukket.
- Fjern forsigtigt prøverne uden at ryste og observere under lys. Et klart hvidt bånd på 95 % rene kerner skal være synligt mellem andet og tredje lag (figur 1).
4. Isolering af kerner
- Aspirere de øverste lag, indtil den hvide kernebånd på interfase af 29%-35%.
- Kernebåndet opsamles i et 200 mL-volumen, overføres til et friskrør og filtreres med et 20 μm filter (Materialetabel).
BEMÆRK: Kernerne behøver ikke at blive suspenderet igen før filtrering. - Kontroller under et let mikroskop for at kontrollere fjernelsen af store snavs og den nukleare membrans intakthed. Kerner skal være runde, og atommembranen må ikke forvrænges.
- Tæl kerner ved hjælp af Trypan blå farvning på et hæmocytometer og aliquot kerner til snRNA-seq/snATAC-seq.
5. Enkelt kerner RNA og ATAC seq
- Proces straks kernerne ved hjælp af enkeltcellegenekspression og ENKELTcellede ATAC-reagenssæt (Materialetabel).
BEMÆRK: Kerneprøvekoncentrationerne for 10x Genomforskningssystemet er 1500-3000 kerner pr. μL for snRNA-seq og 3500-7000 kerner pr. m μL for snATAC-seq. Kernerne kan fortyndes ved hjælp af 1x PBS. - Sekvens de resulterende biblioteker på Genomforskning Core Facility.
- Udfør kvalitetskontrolanalyse af dataene. Kerner indgår til yderligere analyse, hvis de indeholder Unique Molecular Identifier (UMI) >1000, antallet af gener >500 og procent af mitokondriegener <5%, og er inden for middelværdi + tre standardafvigelser af UMI'er og gener.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Single nucleus genomforskning er et område i udvikling med begrænsede data og protokoller. En kritisk faktor, der påvirker resultatet af enkelt kerner assays er isolering af ren og intakt kerner. Vi kombinerede to offentliggjorte protokoller (DroNc-seq og Omni-ATAC-seq protokoller) for at isolere høj kvalitet og ren kerner fra friske frosne gliom væv blokke på relativt kort tid og dermed opretholde stabiliteten af udskrifter (Figur 1).
Brugen af forskellige filtreringstrin sammen med gradientcentrifugationen ved hjælp af joxanol/saccharosegradient giver mulighed for isolering af rene kerner, hvor størstedelen af vragrester kasseres (Figur 2). Det samme isolerede kernepræparat kan bruges til både snRNA-seq og snATAC-seq. Det er vigtigt, at da de anvendte kerner er fra det samme eksempel, kan de genererede data integreres sammen ved hjælp af pakker som Seurat til at generere klynger og give et multi-omics perspektiv25 (Figur 3).
For at afgøre, om protokollen kan sammenlignes med offentliggjorte snRNA-seq datasæt, sammenlignede vi data opnået ved hjælp af proceduren med fire offentligt tilgængelige snRNA-seq-undersøgelser relateret til centralnervesystemet (CNS): Slyper et al.20, Lake et al.26, Jakel et al.24 og Habib et al.18. For at sammenligne kvalitetskontrolmålingerne downloadede vi følgende tællematricer fra Gene Expression Omnibus (GEO): GSE104525 (Habib datasæt, 2017), GSE97930 (Sødatasæt, 2018), GSE118257 (Jakel datasæt, 2019) og GSE140819 (Slyper datasæt, 2020). For Lake datasæt, en fælles rå tælle matrix blev skabt ved at fusionere de enkelte matricer for lillehjernen halvkugle, frontal cortex og visuel cortex. For Slyper-datasættet blev der valgt råtælling for prøven HTAPP-443-SMP-5491 (pædiatrisk gliom af høj kvalitet).
For at udføre en upartisk sammenligning blev alle prøver undtagen Jakel-datasæt behandlet ved hjælp af en fælles standardiseret protokol. Først brugte vi Seurat R-statistisk pakke til at skabe en Seurat objekt af hver rå matrix27. Dette blev efterfulgt af to trin: 1) for at fjerne potentielle dråber, følgende cutoffs blev brugt - kerner indeholdende mindre end 1000 UMI, mindre end 500 gener eller mere end 5% af mitokondrie RNA blev udelukket fra analysen og 2) for at udelukke outliers, kerner, der faldt uden for gennemsnittet plus tre standardafvigelser for distribution af UMI'er og gener blev fjernet. For Jakel-datasættet kunne dette andet trin ikke udføres, da det offentligt tilgængelige datasæt blev forbehandlet med et mindre stringent kvalitetskontroltrin.
For at sammenligne fordelingen af UMI'er og gener på tværs af prøver fusionerede vi alle datasæt og visualiserede fordelingen af antallet af UMI'er og gener ved hjælp af et violinplot (Figur 4). Dette resultat viste , at metoden kan sammenlignes med den seneste snRNA-seq-protokol, der er beskrevet i Slyper et al.20.
Protokollen illustreret her beskæftiger sig med gliom prøver, men den samme tilgang kan feaably anvendes for ikke-CNS tumorer og væv. Ikke desto mindre vil dette kræve optimering af lysis buffersammensætninger og inkubationstider.
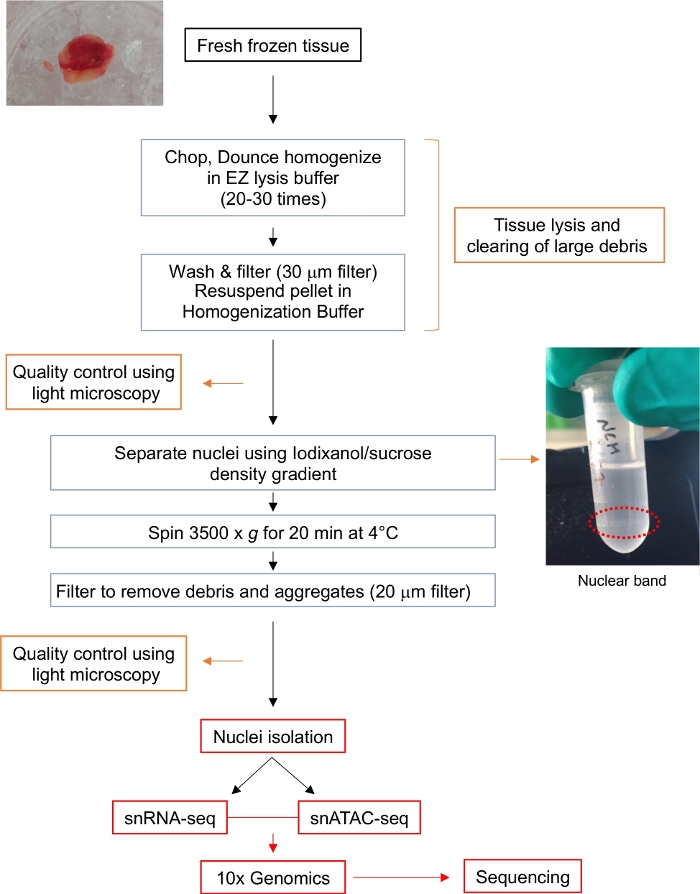
Figur 1: Rutediagram for kernei isolation. Flowdiagrammet giver et kort syn på de trin, der er involveret i isolering af enkelt kerner fra en frisk frossen gliom væv. Repræsentative billeder til tumorprøven og atombåndet efter Iodixanol/saccharosegradient (omgivet af rød prikket linje) vises. Klik her for at se en større version af dette tal.
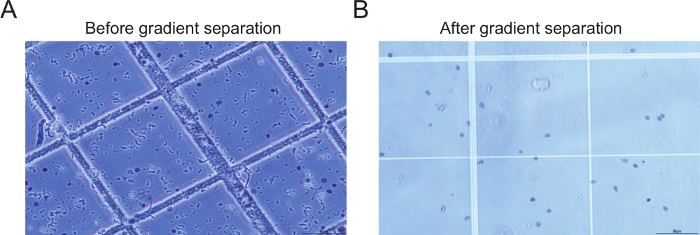
Figur 2: Eksempler på kerner før og efter gradientcentrifugering. (A) Prøvens billede, før gradientcentrifugering og filtrering udføres, viser store mængder snavs (B) Prøvens billede efter gradientcentrifugerings- og filtreringstrin viser intakt kerner med minimal mængde snavs. Klik her for at se en større version af dette tal.

Figur 3: Eksempel på ko-integrerede data fra snRNA-seq og snATAC-seq ved hjælp af samme kernepræparat. Seurat R-statistiske pakke blev brugt til at integrere snRNA-seq- og snATAC-seq-data27. (A) Co-indlejret billede af snRNA-seq og snATAC-seq data (B) Klynger produceret ved co-indlejring af snRNA-seq og snATAC-seq data. Klik her for at se en større version af dette tal.

Figur 4: Kvalitetskontrolparametre for forskellige datasæt for menneskehjerne snRNA-seq, der viser det individuelle antal UMI (A) og antallet af gener (B) pr. kerner. Antallet af kerner, der passerede kvalitetsfiltre, var som følger: 3527 kerner fra Slyper-datasættet, 14636 kerner fra Narayanan-datasættet genereret ved hjælp af den beskrevne protokol, 7369 kerner fra Jakel-datasættet, 16494 kerner fra søens datasæt og 4652 kerner fra Habib-datasættet. Klik her for at se en større version af dette tal.
| 6x homogenisering buffer stabil master mix | ||
| Reagens | Sidste konc. | Vol. for 100 (mL) |
| 1 M CaCl2 | 30 mM | 3.0 |
| 1 m mg(ac)2 | 18 mM | 1.8 |
| 1 M Tris pH 7,8 | 60 mM | 6.0 |
| H2O | 89.2 | |
| Hold ved stuetemperatur, undgå direkte udsættelse for lys | ||
Tabel 1: Udarbejdelse af 6x homogenisering buffer stabil master mix.
| 1 M Saccharose |
| 34,23 g saccharose |
| Opløs i 78,5 mL vand |
| Fyld op til 100 mL med vand |
Tabel 2: Forberedelse af 1 M saccharose.
| 6x homogeniseringsbuffer ustabil løsning (650 gtL pr. prøve) | ||
| Reagens | Sidste konc. | Vol pr. prøve (μL) |
| 6x homogeniseringsbuffer stabil | 6x | 648.84 |
| 100 mM PMSF (Phenylmethylsulfonylfluorid) | 0,1 mM | 1.08 |
| 14,3 M β-mercaptoethanol | 1 mM | 0.08 |
Tabel 3: Fremstilling af 6x homogeniseringsbuffer ustabil opløsning (650 μL pr. prøve).
| 1x homogeniseringsbuffer ustabil løsning (2 mL pr. prøve) | ||
| Reagens | Sidste konc. | Vol pr. prøve (μL) |
| 6x homogeniseringsbuffer ustabil | 1x | 333.33 |
| 1 M Saccharose | 320 mM | 640.00 |
| 50 mM EDTA | 0,1 mM | 4.00 |
| 10% NP40 | 0.1% | 20.00 |
| H2O | 1006.27 | |
Tabel 4: Forberedelse af 1x homogeniseringsbuffer ustabil opløsning (2 mL pr. prøve).
| 50% joxanolopløsning (200 μL pr. prøve) | ||
| Reagens | Sidste konc. | Vol pr. prøve (μL) |
| 6x homogeniseringsbuffer ustabil | 1x | 66.67 |
| 60% joxanolopløsning | 50% | 333.33 |
Tabel 5: Fremstilling af 50% joodixanolopløsning (200 μl pr. prøve).
| 29% joxanolopløsning (300 μL pr. prøve) | ||
| Reagens | Sidste konc. | Vol pr. prøve (μL) |
| 6x homogeniseringsbuffer ustabil | 1x | 100 |
| 1 M Saccharose | 160 mM | 96 |
| 60% joxanolopløsning | 29% | 290 |
| H2O | 114 | |
Tabel 6: Fremstilling af 29% joodixanolopløsning (300 μl pr. prøve).
| 35% joxanolopløsning (300 μL pr. prøve) | ||
| Reagens | Sidste konc. | Vol pr. prøve (μL) |
| 6x homogeniseringsbuffer ustabil | 1x | 100 |
| 1 M Saccharose | 160 mM | 96 |
| 60% joxanolopløsning | 35% | 350 |
| H2O | 54 | |
Tabel 7: Fremstilling af 35% joodixanolopløsning (300 μl pr. prøve).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Inden for intra-tumoral heterogenitet er på et spændende stadium, med nye analyser og platforme, der udvikles til at udfordre og udvide den eksisterende viden. Intra-tumoral heterogenitet er en afgørende faktor, der bidrager til sygdomsprogression og resistens over for de nuværende behandlingsmetoder i gliomer28. Nylige undersøgelser af hjernetumorer har fokuseret på dette vigtige aspekt ved hjælp af enkeltcellede transskriptoriske og epigenomiske analyser for bedre at karakterisere den cellulære heterogenitet inden for samme tumor29,30,31,32. En af de nuværende flaskehalse med enkeltcelleanalyser i hjernetumorer, og andre solide tumorer, er tilgængeligheden af friske kliniske prøver. For at overvinde dette problem har forskellige undersøgelser vist, at brug af kerner fra frisk frosset væv kan være et alternativ til friske prøver og kan med succes bruges til at afhøre cellulær heterogenitet18,33.
Her forbedrer vi de tidligere enkeltkernei isolationsmetoder med hensyn til enkelhed, længde, overflod og kvalitet af kerner. Denne tilgang yderligere bringer fordelen ved profilering snRNA-seq og snATAC-seq fra samme kerner forberedelse, giver mulighed for multi-omics undersøgelser. I denne procedure har vi ændret eksisterende protokoller ved at tilføje en ekstra proces med gradientadskillelse ved hjælp af et ikke-ionisk iodixanol-baseret medium. Denne tilgang giver mulighed for isolering af rene kerner uden behov for FACS sortering, og kvaliteten af kernerne vurderes under et let mikroskop. En god kvalitet suspension af kerner bør ikke have klumpning, minimal snavs, og intakt kerner. Selvom det er sjældent, kan klumpning af kerner forekomme under isolationstrinnet, som kan løses ved let pipetter ved hjælp af bredborende P200 pipettespidser eller ved at belaste kernerne gennem et 20 mm filter. Den mest kritiske faktor i enkelt kerne genomforskning er evnen til at opnå ren og intakt enkelt kerner. Tilstedeværelsen af snavs eller aggregater kan føre til en blok af de mikrofluidiske kamre inden for enkeltcelleplatformene, hvilket fører til enten data af lav kvalitet eller til en mulig fejl i eksperimentet. Formålet med at anvende filtre på forskellige trin efterfulgt af observation under et lysmikroskop er at forhindre sådanne hændelser.
Mængden og kvaliteten af udgangsmateriale (f.eks frisk frossen tumor) er også vigtige overvejelser. Vi har med succes brugt den protokol, der er beskrevet her med hjernetumorvæv blokke spænder fra 10 mg til 60 mg. For disse prøver er 500 μL kerner af nuclei lysis buffer (trin 1) tilstrækkelig til at opnå tilstrækkelig kerner af god kvalitet til enkeltkerne sekventering. Betydeligt større tumorblokke, der indeholder mere end 60-70% tumorindhold, kan hakkes i mindre stykker, og stykker kan enten straks placeres tilbage i flydende nitrogen eller -80 °C opbevaring eller opdeles i rør til enkelt kerneiisolering og blandes, når lysis-trinnet er afsluttet. Derudover bør vævskvalitet og tumorindhold vurderes af Hæmatoxylin og Eosin (H&E) farvning efterfulgt af bekræftelse fra en patolog.
De næste vigtige faktorer at overveje er temperatur og behandlingstid. Forkert vævslagring, prøvehåndtering og lange protokoller kan have en negativ indvirkning på kvaliteten af de endelige genomdata. Prøverne skal opbevares på is, og tiden for prøvebehandling skal være hurtig for at forhindre nedbrydning af kerner. Den gennemsnitlige behandling for en prøve med denne protokol er 45-60 minutter, og det tager cirka 2 timer at behandle fire prøver. Forkert vævsopbevaring kan også påvirke kerneiisolering og datakvalitet. Vi vurderede, om lagringstidens længde ved -80 °C påvirkede kvaliteten af de data, der genereres ved hjælp af den beskrevne protokol. Sammenligningen af sekventeringsresultaterne fra de frosne prøver, der blev banket mellem 1 og 14 år (2006-2019), viste ingen forskelle i datakvaliteten med hensyn til opbevaringstid. Derfor kan korrekt frosne og lagrede friske frosne vævsprøver bruges til retrospektive undersøgelser ved hjælp af denne protokol.
Der er flere begrænsninger med de enkelte kerneprotokoller. Forskellige celletyper kan f.eks. ikke sorteres ud fra celleoverflademarkører, og cytoplasmaiske udskrifter kan ikke påvises. Vi udelader også mitokondrieudskrifter og går derfor glip af visse biologiske oplysninger relateret til tumormetabolisme. Desuden kan forskellige vævstyper kræve forskellige lysis buffere og dermed kræve optimering af protokollen med hver ny eksperimentel opsætning.
Samlet set er den protokol, der er beskrevet her, enkel, reducerer prøvebehandlingstiden og giver kerner af høj kvalitet. Manglen på et sorteringstrin reducerer stress på kernerne og eliminerer behovet for en cellesortering. Fraværet af mitokondrie RNA øger sekventering dybden af nukleare udskrifter. Det er vigtigt, at denne forbedrede protokol i lighed med andre enkeltkerneiisolationsprotokoller letter brugen af arkiverede frosne prøver til retrospektiv analyse. Vi har med succes anvendt denne arbejdsgang til at udføre snRNA-seq og snATAC-seq ved hjælp af det samme kernepræparat fra hjernetumorer uden behov for at ændre buffere. Derfor giver denne tilgang mulighed for multi-omics undersøgelser fra samme prøve.
Afslutningsvis er den enkelte kernei isolationsmetode, der er beskrevet her, en meget effektiv, præcis og hurtig teknik, som kan anvendes til at udføre sekventeringsundersøgelser med en enkelt kerne.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne har intet at afsløre.
Acknowledgments
Vi takker Single Cell Open Lab (scOpenLab) på German Cancer Center (DKFZ) for nyttige diskussioner. Denne forskning blev støttet af den tyske Cancer Aid, Max-Eder Program tilskud nummer 70111964 (S.T.).
Materials
| Name | Company | Catalog Number | Comments |
| 2-Mercaptoethanol | Sigma | M6250 | |
| CaCl2 | Sigma | 21115-100ML | |
| Dounce Homogenizer | Active motif | 40401 | |
| EDTA (0.5 M) | Thermo Scientific | R1021 | |
| Falcon 15 mL Conical Centrifuge Tubes | Fisher Scientific | 352096 | |
| Iodixanol (aka Optiprep) | Stem cell technologies | 07820 | |
| MACs Smart Strainers (30 µm) | Miltenyi Biotec | 130-098-458 | |
| MACS SmartStrainers (100 µm) | Miltenyi Biotec | 130-098-463 | |
| Mg(Ac)2 | Sigma | 63052-100ML | |
| NP-40 | Abcam | ab142227 | |
| Nuclei Isolation Kit: Nuclei EZ Prep | Sigma | NUC101-1KT | |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma | P7626 | |
| Pre-Separation Filters (20 µm) | Miltenyi Biotec | 130-101-812 | |
| Safe lock tubes 1.5 mL | Eppendorf | 0030120086 | |
| Safe lock tubes 2.0 mL | Eppendorf | 0030120094 | |
| Single Cell ATAC | 10x Genomics | ||
| Single Cell Gene Expression | 10x Genomics | ||
| Sucrose | Sigma | S0389 | |
| Wide Bore pipette tips (1000 µL) | Themo Fisher Scientific | 2079GPK | |
| Wide Bore pipette tips (200 µL) | Themo Fisher Scientific | 2069GPK |
References
- Huse, J. T., Holland, E. C. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature Reviews Cancer. 10 (5), 319-331 (2010).
- Kreso, A., Dick, J. E. Evolution of the cancer stem cell model. Cell Stem Cell. 14 (3), 275-291 (2014).
- Filbin, M. G., Suva, M. L. Gliomas Genomics and Epigenomics: Arriving at the Start and Knowing It for the First Time. Annual Review of Pathology. 11, 497-521 (2016).
- Ferris, S. P., Hofmann, J. W., Solomon, D. A., Perry, A. Characterization of gliomas: from morphology to molecules. Virchows Archive. 471 (2), 257-269 (2017).
- Louis, D. N., et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathologica. 131 (6), 803-820 (2016).
- Eckel-Passow, J. E., et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. The New England Journal of Medicine. 372 (26), 2499-2508 (2015).
- Suzuki, H., et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nature Genetics. 47 (5), 458-468 (2015).
- Cancer Genome Atlas Research. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. The New England Journal of Medicine. 372 (26), 2481-2498 (2015).
- Noushmehr, H., et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 17 (5), 510-522 (2010).
- Yan, H., et al. IDH1 and IDH2 mutations in gliomas. The New England Journal of Medicine. 360 (8), 765-773 (2009).
- Yan, H., Bigner, D. D., Velculescu, V., Parsons, D. W. Mutant metabolic enzymes are at the origin of gliomas. Cancer Research. 69 (24), 9157-9159 (2009).
- Gawad, C., Koh, W., Quake, S. R. Single-cell genome sequencing: current state of the science. Nature Reviews Genetics. 17 (3), 175-188 (2016).
- Tanay, A., Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature. 541 (7637), 331-338 (2017).
- Wu, M., Singh, A. K. Microfluidic Flow Cytometry for Single-Cell Protein Analysis. Methods in Molecular Biology. 1346, 69-83 (2015).
- Schwartzman, O., Tanay, A. Single-cell epigenomics: techniques and emerging applications. Nature Reviews Genetics. 16 (12), 716-726 (2015).
- Macaulay, I. C., Ponting, C. P., Voet, T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends in Genetics. 33 (2), 155-168 (2017).
- Buenrostro, J. D., et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 523 (7561), 486-490 (2015).
- Habib, N., et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods. 14 (10), 955-958 (2017).
- Corces, M. R., et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature Methods. 14 (10), 959-962 (2017).
- Slyper, M., et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nature Medicine. 26 (5), 792-802 (2020).
- Mathys, H., et al. Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 570 (7761), 332-337 (2019).
- Krishnaswami, S. R., et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature Protocols. 11 (3), 499-524 (2016).
- Al-Dalahmah, O., et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathologica Communications. 8 (1), 19 (2020).
- Jakel, S., et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 566 (7745), 543-547 (2019).
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E., Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. , (2018).
- Lake, B. B., et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nature Biotechnology. 36 (1), 70-80 (2018).
- Stuart, T., et al.
- Bready, D., Placantonakis, D. G.
- Venteicher, A. S., et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 355 (6332), (2017).
- Tirosh, I., et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 539 (7628), 309-313 (2016).
- Weng, Q., et al. Single-Cell Transcriptomics Uncovers Glial Progenitor Diversity and Cell Fate Determinants during Development and Gliomagenesis. Cell Stem Cell. 24 (5), 707-723 (2019).
- Neftel, C., et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 178 (4), 835-849 (2019).
- Al-Ali, R., et al. Single-nucleus chromatin accessibility reveals intratumoral epigenetic heterogeneity in IDH1 mutant gliomas. Acta Neuropathologica Communications. 7 (1), 201 (2019).
