

Summary
Intratumorale Heterogenität ist ein inhärentes Merkmal von Tumoren, einschließlich Gliomen. Wir haben ein einfaches und effizientes Protokoll entwickelt, das eine Kombination aus Puffern und Gradientenzentrifugation verwendet, um einzelne Kerne aus frischem gefrorenem Gliomgewebe für Einzelkern-RNA- und ATAC-Sequenzierungsstudien zu isolieren.
Abstract
Adulte diffuse Gliome weisen Inter- und Intratumorheterogenität auf. Bis vor kurzem konzentrierte sich die Mehrheit der groß angelegten molekularen Profiling-Bemühungen auf Massenansätze, die zur molekularen Klassifizierung von Hirntumoren führten. In den letzten fünf Jahren haben Einzelzellsequenzierungsansätze mehrere wichtige Merkmale von Gliomen hervorgehoben. Die meisten dieser Studien haben frische Hirntumorproben verwendet, um einzelne Zellen mit Durchflusszytometrie oder Antikörper-basierten Trennmethoden zu isolieren. In Zukunft wird die Verwendung von frisch gefrorenen Gewebeproben aus Biobanken eine größere Flexibilität für Einzelzellanwendungen bieten. Darüber hinaus wird die nächste Herausforderung mit dem Fortschreiten des Einzelzellfeldes darin bestehen, Multi-Omics-Daten entweder aus einer einzelnen Zelle oder derselben Probenvorbereitung zu generieren, um die Tumorkomplexität besser zu entschlüsseln. Daher werden einfache und flexible Protokolle, die die Datengenerierung für verschiedene Methoden wie Single-Nucleus-RNA-Sequenzierung (snRNA-seq) und Single-Nucleus-Assay für Transposase-Accessible Chromatin mit Hochdurchsatz-Sequenzierung (snATAC-seq) ermöglichen, für das Feld wichtig sein.
Jüngste Fortschritte im Einzelzellbereich in Verbindung mit zugänglichen mikrofluidischen Instrumenten wie der 10-fachen Genomik-Plattform haben Einzelzellanwendungen in Forschungslabors erleichtert. Um die Heterogenität von Hirntumoren zu untersuchen, haben wir ein verbessertes Protokoll zur Isolierung einzelner Kerne aus frischen gefrorenen Gliomen entwickelt. Dieses Protokoll führt bestehende Einzelzellprotokolle zusammen und kombiniert einen Homogenisierungsschritt, gefolgt von Filtration und puffervermittelter Gradientenzentrifugation. Die resultierenden Proben sind reine Einzelkernsuspensionen, die verwendet werden können, um Einzelkern-Genexpression und Chromatin-Zugänglichkeitsdaten aus derselben Kernpräparation zu generieren.
Introduction
Diffuse niedriggrade Gliome (LGG), der häufigste primäre Hirntumor bei Erwachsenen, sind infiltrative Neoplasmen, die häufig in der Gehirnhemisphäre auftreten. LGGs weisen sowohl inter- als auch intratumorale Heterogenität auf, die nicht nur von der Tumorpopulation, sondern auch von den nicht-malignen Zellen angetriebenwird,die an der Tumorentwicklung und -progression beteiligt sind1,2,3,4,5.
In den letzten zehn Jahren gab es eine Lawine genomischer Daten, die im Bereich der Gliome gesammelt wurden. Diese Daten stammen hauptsächlich aus Massentumorsequenzierungsstudien und haben immens zur molekularen Charakterisierung und der aktuellen Klassifizierung von Hirntumoren5,6,7,8,9,10,11beigetragen . Obwohl diese Studien die breite molekulare Landschaft im Zusammenhang mit Gliomen aufzeigten, gibt es immer noch einen enttäuschenden Mangel an Fortschritten bei der therapeutischen Intervention. Eines der Hindernisse für die Behandlungsresistenz bei Hirntumoren ist die Intratumorheterogenität. Um dieses Problem anzugehen, haben sich verschiedene Studien auf die genomische, transkriptomische, proteomische und epigenetische Heterogenität konzentriert, die in einem Tumor auf Einzelzellebene vorhanden ist12,13,14,15,16,17.
Obwohl es in den letzten Jahren bemerkenswerte technologische Fortschritte im Einzelzellbereich gegeben hat, ist einer der wichtigsten einschränkenden Faktoren die Verfügbarkeit frischer Proben, die benötigt werden, um die Zellen zu isolieren und diese Experimente durchzuführen. Um diese Einschränkung zu überwinden, gab es mehrere erfolgreiche Versuche, Assays wie snRNA-seq und snATAC-seq aus gefrorenem Gewebe durchzuführen, wobei Kerne anstelle von Zellen verwendet wurden18,19. Die meisten dieser Methoden basieren entweder auf fluoreszenzaktivierter Zellsortierung (FACS) oder Filtrationsstrategien. Sowohl Einzelzell- als auch Einzelkernansätze haben ihre Stärken und Nachteile. Einzelzellansätze pflegen mitochondriale Transkripte, die zwar informativ sein können, aber aufgrund ihrer hohen Häufigkeit auch die Transkriptomabdeckung reduzieren können. Einzelkernisolationsansätze eliminieren einen hohen Prozentsatz der mitochondrialen Fraktion, wodurch eine tiefergehende Abdeckung der Kerntranskripte ermöglichtwird 20.
Es gibt verschiedene kommerziell verfügbare Plattformen, die in den letzten Jahren verwendet wurden, um Einzelzellgenomikdaten zu untersuchen, einschließlich RNA-seq und ATAC-seq. Eine der bekanntesten Plattformen ist die 10x Genomics Chromium-Plattform für einzellige Genexpression und Einzelzell-ATAC-Profiling. Da die Plattform mit Hilfe von mikrofluidischen Kammern arbeitet, können Ablagerungen oder Aggregate das System verstopfen, was zum Verlust von Daten, Reagenzien und wertvollen klinischen Proben führt. Daher hängt der Erfolg von Einzelzellstudien weitgehend von der genauen Isolierung einzelner Zellen/Kerne ab.
Das Protokoll, das wir hier demonstrieren werden, ist eine leicht modifizierte Kombination der Protokolle DroNc-seq und Omni-ATAC-seq und verwendet einen ähnlichen Ansatz wie neuere Studien, die snRNA-seq verwenden, um neurologische Störungen und neuronale Zelltypen im menschlichen Gehirn zu verstehen18,19,21,22,23,24. Das Protokoll verwendet eine Kombination aus enzymatischer /mechanischer Dissoziation gefrorener Proben, gefolgt von Filtration und Gradientenzentrifugation und ermöglicht eine schnelle und genaue Isolierung einzelner Kerne aus frischem gefrorenem Gliomgewebe. Wir haben dieses Protokoll erfolgreich verwendet, um snRNA-seq- und snATAC-seq-Daten aus dem gleichen Kernpräparat aus Hirntumorproben zu generieren.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Frische gefrorene Gliomproben wurden aus der Gewebebank des Nationalen Zentrums für Tumorerkrankungen (NCT) in Heidelberg gewonnen. Die Verwendung von Patientenmaterial wurde vom Institutional Review Board der Medizinischen Fakultät Heidelberg genehmigt und die Einwilligung aller in die Studie einbezogenen Patienten eingeholt.
1. Experimentelle Vorbereitung
- Führen Sie alle Schritte auf Eis oder bei 4 °C durch.
- Vorkühlschläuche, Geschirr, Rasierklingen, Douncer und Stößel auf 4 °C.
- Bereiten Sie alle Puffer im Voraus vor. Diese Puffer sind bei Raumtemperatur stabil. Sterilfiltration wird empfohlen, insbesondere für Saccharose. Die Stoffpräparate sind modifiziert von Corces et al.19. Siehe Tabellen 1-7.
- Entfernen Sie Proben aus flüssigem Stickstoff oder -80 °C Gefrierfach und halten Sie sie bis Schritt 2.1 auf Trockeneis.
2. Gewebedissektion und Dissoziation
- Frische gefrorene Gewebeprobe (10-60 mg) in eine vorgekühlte Petrischale geben. Frisches gefrorenes Tuch mit einer Rasierklinge in kleine Stücke auf Eis hacken/ hacken.
- 500 μL gekühlter Kernlysepuffer werden in ein vorgekühltes 1,5 ml Röhrchen gegeben. Legen Sie die Gewebestücke in das 1,5-ml-Röhrchen, das den Kernlysepuffer enthält, und geben Sie es in einen Douncer (Materialtabelle).
HINWEIS: Es gibt zwei Dounce Homogenisatorstößel: "lose" oder "A" Stößel für die anfängliche Probenreduktion und "tight" oder "B" Stößel für die vollständige Probenhomogenisierung. - Verdoppeln Sie die Gewebestücke mit dem "losen" Stößel für etwa 20 Hübe, bis die Reibung reduziert ist. Wenn Ablagerungen vorhanden sind, kann die Probe durch Filtration mit einem 100-mm-Sieb vorgeräumt werden.
- Dounce mit dem "engen" Stößel für 20 Schläge, um eine vollständige Gewebehomogenisierung zu erreichen.
- Das Homogenat (ca. 500 μL) wird in ein vorgekühltes 2 mL Röhrchen überführen. Fügen Sie 1 ml gekühlten Lysepuffer hinzu. Sanft mischen und 5 min auf Eis inkubieren. Vorsichtig mit einer Breitrohrpipettenspitze mischen und während der Inkubation 1-2 mal wiederholen.
- Filtern Sie das gesamte Homogenat mit einem 30 μm Siebgewebe, sammeln Sie es in einem 15 mL Falcon Rohr und geben Sie es wieder in ein neues vorgekühltes 2 mL Rohr um. Ein einzelnes Sieb reicht typischerweise für das gesamte Homogenat aus.
- Überprüfen Sie unter einem Lichtmikroskop, um die Entfernung großer Ablagerungen und die Unversehrtheit der Kernmembran zu überprüfen. Kerne müssen rund sein und die Kernmembran sollte nicht verzerrt werden. Wenn Ablagerungen vorhanden sind, können Kerne erneut gefiltert werden.
- Zentrifuge die Kerne bei 500 x g für 5 min bei 4 °C auf einer Tischzentrifuge. Entfernen Sie den Überstand und lassen Sie ~ 50 μL mit Pellet zurück, das die Kerne enthält. Das Pellet vorsichtig in einem weiteren 1 ml Kernlysepuffer resuspendieren und 5 min auf Eis inkubieren.
- Zentrifuge die Kerne bei 500 x g für 5 min bei 4°C. Entfernen Sie den Überstand ohne störendes Pellet, fügen Sie 500 ml 1x Homogenisierungspuffer (HB) hinzu (Tabelle 4) und inkubieren Sie für 5 min ohne Wiederaufwirbeln. Dann resuspendieren Sie die Kerne in einem weiteren 1,0 ml 1x HB.
- Zentrifuge die Kerne bei 500 x g für 5 min bei 4 °C. Entfernen Sie den Überstand und resuspendieren Sie die Kerne vorsichtig in 200 μL 1x HB in ein neues 2,0 ml Röhrchen.
3. Gradientenzentrifugation
- Fügen Sie 200 μL 50% Iodixanollösung (Tabelle 5) hinzu, um eine Endkonzentration von 25% Iodixanol zu erhalten. 10 mal mit Pipette auf 300 ml gut mischen.
- 300 μL 29% ige Iodixanollösung (Tabelle 6) werden unter die 25% ige Mischung geben. Verwenden Sie eine P1000-Feinspitze, um eine Vermischung der Schichten zu vermeiden.
- 300 μl 35%ige Iodixanollösung (Tabelle 7) werden unter die 29%ige Mischung geben. Verwenden Sie eine P1000-Feinspitze, um eine Vermischung der Schichten zu vermeiden.
Achtung: Dieser Schritt erfordert eine schrittweise Entfernung der Pipettenspitze während des Pipettierens, um eine übermäßige Volumenverschiebung zu vermeiden. - Legen Sie die Proben in eine Schwenkschaufelzentrifuge, drehen Sie sie 20 min bei 3.500 x g bei 4°C mit ausgeschalteter Bremse.
- Entfernen Sie die Proben vorsichtig ohne Zuschütteln und beobachten Sie sie unter Licht. Zwischen der zweiten und dritten Schicht sollte ein klares weißes Band aus 95% reinen Kernen sichtbar sein (Abbildung 1).
4. Isolierung von Kernen
- Saugen Sie die oberen Schichten an, bis sich die weißen Kerne in der Interphase von 29% -35% banden.
- Sammeln Sie das Kernband in einem Volumen von 200 ml, geben Sie es in ein frisches Rohr und filtern Sie es mit einem 20 μm Filter (Materialtabelle).
HINWEIS: Die Kerne müssen vor der Filtration nicht wieder aufgewendet werden. - Überprüfen Sie unter einem Lichtmikroskop, um die Entfernung großer Ablagerungen und die Unversehrtheit der Kernmembran zu überprüfen. Kerne müssen rund sein und die Kernmembran sollte nicht verzerrt werden.
- Zählen Sie Kerne mit Trypan-Blaufärbung auf einem Hämozytometer und aliquoten Kernen für snRNA-seq/snATAC-seq.
5. Einzelkern-RNA und ATAC seq
- Sofortige Verarbeitung der Kerne unter Verwendung der Einzelzell-Genexpression und einzelliger ATAC-Reagenz-Kits (Materialtabelle).
HINWEIS: Die Kernprobenkonzentrationen für das 10x Genomics-System betragen 1500-3000 Kerne pro μL für snRNA-seq und 3500-7000 Kerne pro m μL für snATAC-seq. Die Kerne können mit 1x PBS verdünnt werden. - Sequenzieren Sie die resultierenden Bibliotheken in der Genomics Core Facility.
- Führen Sie eine Qualitätskontrollanalyse der Daten durch. Kerne werden für die weitere Analyse einbezogen, wenn sie Unique Molecular Identifier (UMI) >1000, die Anzahl der Gene >500 und Prozent der mitochondrialen Gene <5% enthalten und innerhalb des Mittelwerts + drei Standardabweichungen der UMIs und Gene liegen.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Die Einzelkerngenomik ist ein sich entwickelndes Feld mit begrenzten Daten und Protokollen. Ein kritischer Faktor, der das Ergebnis von Einzelkernassays beeinflusst, ist die Isolierung reiner und intakter Kerne. Wir haben zwei veröffentlichte Protokolle (DroNc-seq- und Omni-ATAC-seq-Protokolle) kombiniert, um in relativ kurzer Zeit hochwertige und reine Kerne aus frischen gefrorenen Gliomgewebeblöcken zu isolieren und so die Stabilität der Transkripte zu erhalten (Abbildung 1).
Die Verwendung verschiedener Filtrationsschritte zusammen mit der Gradientenzentrifugation unter Verwendung des Iodixanol/Saccharose-Gradienten ermöglicht die Isolierung reiner Kerne, wobei der Großteil der Trümmer verworfen wird (Abbildung 2). Die gleiche isolierte Kernpräparation kann sowohl für snRNA-seq als auch für snATAC-seq verwendet werden. Da die verwendeten Kerne aus derselben Stichprobe stammen, können die generierten Daten mitHilfe von Paketen wie Seurat co-eingebettet werden, um Cluster zu generieren und eine Multi-Omics-Perspektive bereitzustellen25 (Abbildung 3).
Um festzustellen, ob das Protokoll mit veröffentlichten snRNA-seq-Datensätzen vergleichbar ist, verglichen wir die mit dem Verfahren erhaltenen Daten mit vier öffentlich zugänglichen snRNA-seq-Studien zum zentralen Nervensystem (ZNS): Slyper et al.20, Lake et al.26, Jakel et al.24 und Habib et al.18. Um die Qualitätskontrollmetriken zu vergleichen, haben wir die folgenden Zählmatrizen aus dem Gene Expression Omnibus (GEO) heruntergeladen: GSE104525 (Habib-Datensatz, 2017), GSE97930 (Lake-Datensatz, 2018), GSE118257 (Jakel-Datensatz, 2019) und GSE140819 (Slyper-Datensatz, 2020). Für den Lake-Datensatz wurde eine gemeinsame Rohzählmatrix erstellt, indem die einzelnen Matrizen für die Kleinhirnhemisphäre, den frontalen Kortex und den visuellen Kortex zusammengeführt wurden. Für den Slyper-Datensatz wurde die Rohzählung für die Probe HTAPP-443-SMP-5491 (hochgradiges pädiatrisches Gliom) ausgewählt.
Um einen unvoreingenommenen Vergleich durchzuführen, wurden alle Proben mit Ausnahme des Jakel-Datensatzes mit einem gemeinsamen standardisierten Protokoll verarbeitet. Zuerst haben wir das Seurat R-Statistikpaket verwendet, um ein Seurat-Objekt jeder Rohmatrix27zu erstellen. Es folgten zwei Schritte: 1) Um potenzielle Tröpfchen zu entfernen, wurden die folgenden Cutoffs verwendet - Kerne mit weniger als 1000 UMI, weniger als 500 Gene oder mehr als 5% mitochondrialer RNA wurden von der Analyse ausgeschlossen und 2) um Ausreißer auszuschließen, Kerne, die außerhalb des Mittelwerts lagen, plus drei Standardabweichungen für die Verteilung von UMIs und Genen wurden entfernt. Für den Jakel-Datensatz konnte dieser zweite Schritt nicht durchgeführt werden, da der öffentlich verfügbare Datensatz mit einem weniger strengen Qualitätskontrollschritt vorverarbeitet wurde.
Um die Verteilung von UMIs und Genen auf Proben zu vergleichen, haben wir alle Datensätze zusammengeführt und die Verteilung der Anzahl der UMIs und Gene mit einem Violindiagramm visualisiert (Abbildung 4). Dieses Ergebnis zeigte, dass die Methode mit dem neuesten snRNA-seq-Protokoll vergleichbar ist, das in Slyper et al.20beschrieben wird.
Das hier dargestellte Protokoll befasst sich mit Gliomproben, aber der gleiche Ansatz kann für Nicht-ZNS-Tumoren und Gewebe angewendet werden. Dies erfordert jedoch eine Optimierung der Lysepufferzusammensetzungen und inkubationszeiten.
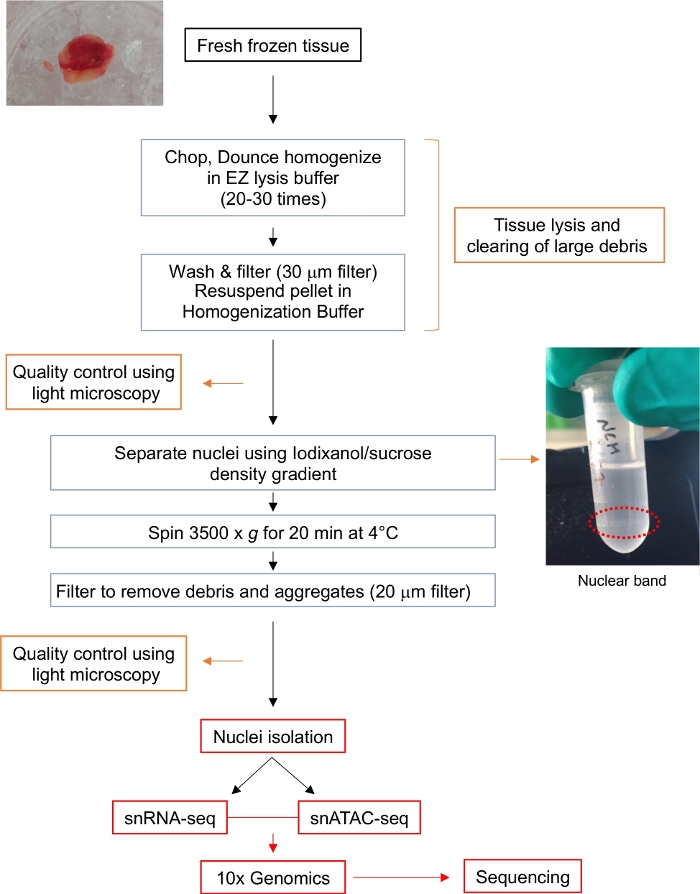
Abbildung 1: Flussdiagramm für die Kernisolierung. Das Flussdiagramm gibt einen kurzen Ausblick auf die Schritte, die bei der Isolierung einzelner Kerne aus einem frischen gefrorenen Gliomgewebe erforderlich sind. Es werden repräsentative Bilder für die Tumorprobe und das Kernband nach Iodixanol/Saccharose-Gradient (eingekreist mit rot gepunkteter Linie) gezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
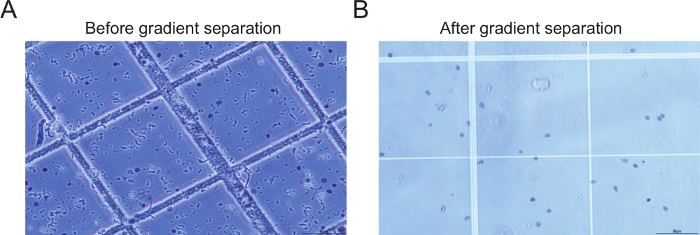
Abbildung 2: Beispiele für Kerne vor und nach der Gradientenzentrifugation. (A) Das Bild der Probe vor der Gradientenzentrifugation und -filtration zeigt große Mengen an Trümmern (B) Das Bild der Probe nach gradientenzentrifugation und Filtrationsschritt zeigt intakte Kerne mit minimaler Menge an Ablagerungen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 3: Beispiel für co-eingebettete Daten aus snRNA-seq und snATAC-seq unter Verwendung derselben Kernpräparation. Das Seurat R-Statistikpaket wurde verwendet, um die snRNA-seq- und snATAC-seq-Daten27zu integrieren. (A) Co-embedded image of snRNA-seq and snATAC-seq data (B) Clusters, die durch co-Embedding von snRNA-seq- und snATAC-seq-Daten erzeugt werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 4: Qualitätskontrollparameter verschiedener snRNA-seq-Datensätze des menschlichen Gehirns, die die individuelle Anzahl von UMI (A) und die Anzahl der Gene (B) pro Kerne zeigen. Die Anzahl der Kerne, die Qualitätsfilter bestanden, war wie folgt: 3527 Kerne aus dem Slyper-Datensatz, 14636 Kerne aus dem Narayanan-Datensatz, die mit dem beschriebenen Protokoll generiert wurden, 7369 Kerne aus dem Jakel-Datensatz, 16494 Kerne aus dem Lake-Datensatz und 4652 Kerne aus dem Habib-Datensatz. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
| 6x Homogenisierungspuffer Stabiler Master Mix | ||
| Reagenz | Abschließende Konk. | Vol für 100 (ml) |
| 1 M CaCl2 | 30 mM | 3.0 |
| 1 m mg(ac)2 | 18 mM | 1.8 |
| 1 M Tris pH 7,8 | 60 mM | 6.0 |
| H2O | 89.2 | |
| Bei Raumtemperatur halten, direkte Lichteinwirkung vermeiden | ||
Tabelle 1: Herstellung von 6x Homogenisierungspuffer stabilem Master Mix.
| 1 M Saccharose |
| 34,23 g Saccharose |
| In 78,5 ml Wasser auflösen |
| Füllen Sie bis zu 100 ml mit Wasser |
Tabelle 2: Zubereitung von 1 M Saccharose.
| 6x Homogenisierungspuffer Instabile Lösung (650 ml pro Probe) | ||
| Reagenz | Abschließende Konk. | Vol pro Probe (μL) |
| 6x Homogenisierungspuffer stabil | 6x | 648.84 |
| 100 mM PMSF (Phenylmethylsulfonylfluorid) | 0,1 mM | 1.08 |
| 14,3 M β-Mercaptoethanol | 1 mM | 0.08 |
Tabelle 3: Herstellung von 6x Homogenisierungspuffer Instabile Lösung (650 μL pro Probe).
| 1x Homogenisierungspuffer Instabile Lösung (2 ml pro Probe) | ||
| Reagenz | Abschließende Konk. | Vol pro Probe (μL) |
| 6x Homogenisierungspuffer Instabil | 1x | 333.33 |
| 1 M Saccharose | 320 mM | 640.00 |
| 50 mM EDTA | 0,1 mM | 4.00 |
| 10% NP40 | 0.1% | 20.00 |
| H2O | 1006.27 | |
Tabelle 4: Herstellung von 1x Homogenisierungspuffer Instabile Lösung (2 ml pro Probe).
| 50% Iodixanol Lösung (200 μL pro Probe) | ||
| Reagenz | Abschließende Konk. | Vol pro Probe (μL) |
| 6x Homogenisierungspuffer Instabil | 1x | 66.67 |
| 60% Iodixanol Lösung | 50% | 333.33 |
Tabelle 5: Herstellung von 50% Iodixanol-Lösung (200 μl pro Probe).
| 29% Iodixanol Lösung (300 μL pro Probe) | ||
| Reagenz | Abschließende Konk. | Vol pro Probe (μL) |
| 6x Homogenisierungspuffer Instabil | 1x | 100 |
| 1 M Saccharose | 160 mM | 96 |
| 60% Iodixanol Lösung | 29% | 290 |
| H2O | 114 | |
Tabelle 6: Herstellung von 29% Iodixanol-Lösung (300 μl pro Probe).
| 35% Iodixanol Lösung (300 μL pro Probe) | ||
| Reagenz | Abschließende Konk. | Vol pro Probe (μL) |
| 6x Homogenisierungspuffer Instabil | 1x | 100 |
| 1 M Saccharose | 160 mM | 96 |
| 60% Iodixanol Lösung | 35% | 350 |
| H2O | 54 | |
Tabelle 7: Herstellung von 35% igen Iodixanollösung (300 μl pro Probe).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Das Gebiet der intratumoralen Heterogenität befindet sich in einem spannenden Stadium, wobei neuartige Assays und Plattformen entwickelt werden, um das vorhandene Wissen herauszufordern und zu erweitern. Intratumorale Heterogenität ist ein entscheidender Faktor, der zum Fortschreiten der Krankheit und zur Resistenz gegen die derzeitigen Behandlungsmodalitäten bei Gliomen beiträgt28. Jüngste Studien zu Hirntumoren haben sich auf diesen wichtigen Aspektkonzentriert,indem sie einzellige transkriptomische und epigenomische Assays verwendet haben, um die zelluläre Heterogenität innerhalb desselben Tumors besser zu charakterisieren29,30,31,32. Einer der aktuellen Engpässe bei den Einzelzellassays in Hirntumoren und anderen soliden Tumoren ist die Verfügbarkeit frischer klinischer Proben. Um dieses Problem zu überwinden, haben verschiedene Studien gezeigt, dass die Verwendung von Kernen aus frischem gefrorenem Gewebe eine Alternative zu frischen Proben sein kann und erfolgreich zur Untersuchung der zellulären Heterogenität verwendet werden kann18,33.
Hier verbessern wir die bisherigen Einzelkernisolationsmethoden in Bezug auf Einfachheit, Länge, Häufigkeit und Qualität der Kerne. Dieser Ansatz bietet außerdem den Vorteil, snRNA-seq und snATAC-seq aus derselben Kernpräparation zu profilieren, was Multi-Omics-Studien ermöglicht. In diesem Verfahren haben wir bestehende Protokolle modifiziert, indem wir einen zusätzlichen Prozess der Gradiententrennung mit einem nichtionischen Iodixanol-basierten Medium hinzugefügt haben. Dieser Ansatz ermöglicht die Isolierung reiner Kerne ohne FACS-Sortierung, und die Qualität der Kerne wird unter einem Lichtmikroskop beurteilt. Eine hochwertige Suspension von Kernen sollte keine Verklumpung, minimale Ablagerungen und intakte Kerne haben. Obwohl selten, kann es während des Isolationsschritts zu Verklumpungen von Kernen kommen, die durch leichtes Pipettieren mit P200-Pipettenspitzen mit breiter Bohrung oder durch Abseihen der Kerne durch einen 20-mm-Filter aufgelöst werden können. Der kritischste Faktor in der Einzelkerngenomik ist die Fähigkeit, reine und intakte Einzelkerne zu erhalten. Das Vorhandensein von Trümmern oder Aggregaten kann zu einem Block der mikrofluidischen Kammern innerhalb der Einzelzellplattformen führen, was entweder zu minderwertigen Daten oder zu einem möglichen Scheitern des Experiments führt. Der Zweck der Verwendung von Filtern in verschiedenen Schritten, gefolgt von der Beobachtung unter einem Lichtmikroskop, besteht darin, solche Vorkommnisse zu verhindern.
Die Menge und Qualität des Ausgangsmaterials (z. B. frisch gefrorener Tumor) sind ebenfalls wichtige Überlegungen. Wir haben das hier beschriebene Protokoll erfolgreich mit Hirntumorgewebeblöcken von 10 mg bis 60 mg angewendet. Für diese Proben reichen 500 μL Kernlysepuffer (Schritt 1) aus, um ausreichend hochwertige Kerne für die Sequenzierung einzelner Kerne zu erhalten. Deutlich größere Tumorblöcke, die mehr als 60-70% Tumorgehalt enthalten, können in kleinere Stücke gehackt werden, und Stücke können entweder sofort wieder in flüssigen Stickstoff oder -80 °C Lagerung gelegt oder in Röhrchen zur Isolierung einzelner Kerne aufgeteilt und gemischt werden, sobald der Lyseschritt abgeschlossen ist. Darüber hinaus sollten gewebequalität und Tumorgehalt durch Hämatoxylin- und Eosin (H & E) -Färbung mit anschließender Bestätigung durch einen Pathologen beurteilt werden.
Die nächsten wichtigen Faktoren, die zu berücksichtigen sind, sind Temperatur und Zeit der Verarbeitung. Unsachgemäße Gewebelagerung, Probenhandhabung und langwierige Protokolle können sich negativ auf die Qualität der endgültigen Genomikdaten auswirken. Die Proben müssen auf Eis gehalten werden, und die Zeit für die Probenverarbeitung muss schnell sein, um den Abbau von Kernen zu verhindern. Die durchschnittliche Verarbeitung für eine Probe mit diesem Protokoll beträgt 45-60 Minuten, und die Verarbeitung von vier Proben dauert etwa 2 Stunden. Eine unsachgemäße Gewebespeicherung kann auch die Isolierung der Kerne und die Datenqualität beeinträchtigen. Wir haben bewertet, ob die Speicherdauer bei -80 °C die Qualität der mit dem beschriebenen Protokoll generierten Daten beeinflusst. Der Vergleich der Sequenzierungsergebnisse aus den eingefrorenen Proben zwischen 1 und 14 Jahren (2006-2019) ergab keine Unterschiede in der Datenqualität in Bezug auf die Lagerzeit. Daher können ordnungsgemäß gefrorene und gelagerte frische gefrorene Gewebeproben für retrospektive Studien mit diesem Protokoll verwendet werden.
Es gibt mehrere Einschränkungen bei den Einzelkernprotokollen. Beispielsweise können verschiedene Zelltypen nicht anhand von Zelloberflächenmarkern sortiert werden, und zytoplasmatische Transkripte sind nicht nachweisbar. Außerdem lassen wir die mitochondrialen Transkripte weg, wodurch bestimmte biologische Informationen im Zusammenhang mit dem Tumorstoffwechsel verloren gehen. Darüber hinaus können verschiedene Gewebetypen unterschiedliche Lysepuffer erfordern, was eine Optimierung des Protokolls mit jedem neuen Versuchsaufbau erfordert.
Insgesamt ist das hier beschriebene Protokoll einfach, reduziert die Probenverarbeitungszeit und liefert hochwertige Kerne. Das Fehlen eines Sortierschritts reduziert die Belastung der Kerne und macht einen Zellsortierer überflüssig. Das Fehlen von mitochondrialer RNA erhöht die Sequenzierungstiefe von Kerntranskripten. Wichtig ist, dass dieses erweiterte Protokoll, ähnlich wie andere Einzelkernisolationsprotokolle, die Verwendung archivierter gefrorener Proben für die retrospektive Analyse erleichtert. Wir haben diesen Workflow erfolgreich angewendet, um snRNA-seq und snATAC-seq mit dem gleichen Kernpräparat aus Hirntumoren durchzuführen, ohne dass Puffer geändert werden müssen. Daher ermöglicht dieser Ansatz Multi-Omics-Studien aus derselben Probe.
Zusammenfassend lässt sich sagen, dass die hier beschriebene Einzelkernisolationsmethode eine hochwirksame, genaue und schnelle Technik ist, die zur Durchführung von Einzelkernsequenzierungsstudien angewendet werden kann.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren haben nichts preiszugeben.
Acknowledgments
Wir danken dem Single Cell Open Lab (scOpenLab) am Deutschen Krebszentrum (DKFZ) für hilfreiche Gespräche. Diese Forschung wurde von der Deutschen Krebshilfe, Max-Eder-Programm Fördernummer 70111964 (S.T.) unterstützt.
Materials
| Name | Company | Catalog Number | Comments |
| 2-Mercaptoethanol | Sigma | M6250 | |
| CaCl2 | Sigma | 21115-100ML | |
| Dounce Homogenizer | Active motif | 40401 | |
| EDTA (0.5 M) | Thermo Scientific | R1021 | |
| Falcon 15 mL Conical Centrifuge Tubes | Fisher Scientific | 352096 | |
| Iodixanol (aka Optiprep) | Stem cell technologies | 07820 | |
| MACs Smart Strainers (30 µm) | Miltenyi Biotec | 130-098-458 | |
| MACS SmartStrainers (100 µm) | Miltenyi Biotec | 130-098-463 | |
| Mg(Ac)2 | Sigma | 63052-100ML | |
| NP-40 | Abcam | ab142227 | |
| Nuclei Isolation Kit: Nuclei EZ Prep | Sigma | NUC101-1KT | |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma | P7626 | |
| Pre-Separation Filters (20 µm) | Miltenyi Biotec | 130-101-812 | |
| Safe lock tubes 1.5 mL | Eppendorf | 0030120086 | |
| Safe lock tubes 2.0 mL | Eppendorf | 0030120094 | |
| Single Cell ATAC | 10x Genomics | ||
| Single Cell Gene Expression | 10x Genomics | ||
| Sucrose | Sigma | S0389 | |
| Wide Bore pipette tips (1000 µL) | Themo Fisher Scientific | 2079GPK | |
| Wide Bore pipette tips (200 µL) | Themo Fisher Scientific | 2069GPK |
References
- Huse, J. T., Holland, E. C. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature Reviews Cancer. 10 (5), 319-331 (2010).
- Kreso, A., Dick, J. E. Evolution of the cancer stem cell model. Cell Stem Cell. 14 (3), 275-291 (2014).
- Filbin, M. G., Suva, M. L. Gliomas Genomics and Epigenomics: Arriving at the Start and Knowing It for the First Time. Annual Review of Pathology. 11, 497-521 (2016).
- Ferris, S. P., Hofmann, J. W., Solomon, D. A., Perry, A. Characterization of gliomas: from morphology to molecules. Virchows Archive. 471 (2), 257-269 (2017).
- Louis, D. N., et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathologica. 131 (6), 803-820 (2016).
- Eckel-Passow, J. E., et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. The New England Journal of Medicine. 372 (26), 2499-2508 (2015).
- Suzuki, H., et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nature Genetics. 47 (5), 458-468 (2015).
- Cancer Genome Atlas Research. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. The New England Journal of Medicine. 372 (26), 2481-2498 (2015).
- Noushmehr, H., et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 17 (5), 510-522 (2010).
- Yan, H., et al. IDH1 and IDH2 mutations in gliomas. The New England Journal of Medicine. 360 (8), 765-773 (2009).
- Yan, H., Bigner, D. D., Velculescu, V., Parsons, D. W. Mutant metabolic enzymes are at the origin of gliomas. Cancer Research. 69 (24), 9157-9159 (2009).
- Gawad, C., Koh, W., Quake, S. R. Single-cell genome sequencing: current state of the science. Nature Reviews Genetics. 17 (3), 175-188 (2016).
- Tanay, A., Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature. 541 (7637), 331-338 (2017).
- Wu, M., Singh, A. K. Microfluidic Flow Cytometry for Single-Cell Protein Analysis. Methods in Molecular Biology. 1346, 69-83 (2015).
- Schwartzman, O., Tanay, A. Single-cell epigenomics: techniques and emerging applications. Nature Reviews Genetics. 16 (12), 716-726 (2015).
- Macaulay, I. C., Ponting, C. P., Voet, T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends in Genetics. 33 (2), 155-168 (2017).
- Buenrostro, J. D., et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 523 (7561), 486-490 (2015).
- Habib, N., et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods. 14 (10), 955-958 (2017).
- Corces, M. R., et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature Methods. 14 (10), 959-962 (2017).
- Slyper, M., et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nature Medicine. 26 (5), 792-802 (2020).
- Mathys, H., et al. Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 570 (7761), 332-337 (2019).
- Krishnaswami, S. R., et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature Protocols. 11 (3), 499-524 (2016).
- Al-Dalahmah, O., et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathologica Communications. 8 (1), 19 (2020).
- Jakel, S., et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 566 (7745), 543-547 (2019).
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E., Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. , (2018).
- Lake, B. B., et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nature Biotechnology. 36 (1), 70-80 (2018).
- Stuart, T., et al.
- Bready, D., Placantonakis, D. G.
- Venteicher, A. S., et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 355 (6332), (2017).
- Tirosh, I., et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 539 (7628), 309-313 (2016).
- Weng, Q., et al. Single-Cell Transcriptomics Uncovers Glial Progenitor Diversity and Cell Fate Determinants during Development and Gliomagenesis. Cell Stem Cell. 24 (5), 707-723 (2019).
- Neftel, C., et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 178 (4), 835-849 (2019).
- Al-Ali, R., et al. Single-nucleus chromatin accessibility reveals intratumoral epigenetic heterogeneity in IDH1 mutant gliomas. Acta Neuropathologica Communications. 7 (1), 201 (2019).
