

Summary
MYTHOS ermöglicht den empfindlichen Nachweis von transienten und stabilen Wechselwirkungen zwischen Proteinen, die im Modellorganismus Saccharomyces cerevisiae exprimiert werden. Es wurde erfolgreich angewendet, um exogene und Hefe integrale Membranproteine zu studieren, um ihre Interaktionspartner in einem hohen Durchsatz Weise zu identifizieren.
Abstract
Die grundlegenden biologischen und klinischen Bedeutung von integralen Membranproteinen veranlasste die Entwicklung einer Hefe-basiertes System für die Hochdurchsatz-Identifizierung von Protein-Protein-Interaktionen (PPI) für full-length Transmembranproteine. Zu diesem Zweck entwickelt unser Labor das Split-Ubiquitin-basierten Membrane Yeast Two-Hybrid (Mythos) System. Diese Technologie ermöglicht den empfindlichen Nachweis von transienten und stabilen Protein-Wechselwirkungen mit
Protocol
1. Hintergrundinformationen
Protein-Protein-Interaktionen (PPI) sind die grundlegenden Bausteine in über alle zellulären Prozesse beteiligt. Daher ist es wichtig, dass alle Interaktionen fest geregelt sind, um zelluläre Homöostase, wie eine Verschiebung in diesem biologische Gleichgewicht im Allgemeinen spielt eine Rolle bei Erkrankungen und Krebs Zelltransformation. Membran-assoziierten Proteine gehören zu den biologisch wichtigen Klasse von Proteinen, wie sie komplexe Signalkaskaden auslösen kann, und vermitteln sowohl den Import und Export verschiedener Moleküle, darunter Medikamente, die der jüngsten Bedeutung im Bereich der Gesundheitsversorgung als Probleme an wurde Resistenzen immer häufiger auf. Der Einblick in die Komplexität dieses Proteins Klasse erfordert die Identifizierung ihrer Interaktionspartner. Die Entdeckung solcher Partner hat sich eine Herausforderung, so oft es erfordert harte Bedingungen, die für jede Membran-gebundenen Proteins optimiert werden muss [1].
Die grundlegenden biologischen und klinischen Bedeutung von integralen Membranproteinen veranlasste die Entwicklung einer Hefe-basiertes System für die Hochdurchsatz-Identifizierung von PPI für full-length Transmembranproteine. Zu diesem Zweck entwickelten wir das Split-Ubiquitin-basierten Membrane Yeast Two-Hybrid (Mythos) System [2-4]. Dieses Tool ermöglicht den empfindlichen Nachweis von transienten und stabilen Protein-Interaktionen. Es wurde erfolgreich eingesetzt, um exogene und endogene Proteine im Modellorganismus Saccharomyces cerevisiae exprimiert [3-7]. Studie MYTHOS nutzt die Beobachtung, dass Ubiquitin in zwei Hälften getrennt werden kann:. Die C-terminale Hälfte (C ub) und der N-terminalen Hälfte (Nubi) in-vivo-Studien haben gezeigt, dass diese Einheiten spontan rekonstruieren aufgrund ihrer hohen Affinität zu ein anderes (Figure. 1a). Allerdings ist die Implementierung einer Isoleucin 13 bis Glycin Punktmutation im N-terminalen Hälfte von Ubiquitin (Herstellung eines Fragments bezeichnet als N ub G) verhindert, dass diese spontane Re-Verein [8] (Figure. 1b).
Wir nutzen dieses Prinzip in der MYTHOS-System (Figure. 1c und 2). Kurz gesagt, ist die integrale Membranproteine Köderprotein eine C ub Gruppierung, die eine künstliche Transkriptionsfaktor, bestehend aus dem Escherichia coli DNA-bindendes Protein LexA und der Aktivierungsdomäne von VP16 von Herpes-simplex-Virus verbunden ist geschmolzen. Preys werden durch Fusion von cDNA oder genomische DNA abgeleitete Fragmente an die NubG Einheit erzeugt. Eine Wechselwirkung zwischen Köder und Beute-Proteine in einem Hefewirt führt zu Wiederherstellung eines full-length "pseudo-Ubiquitin-Molekül, mit anschließender Anerkennung durch cytosolische deubiquitinating Enzyme (DUBs) und proteolytische Freisetzung des Transkriptionsfaktors. Der Transkriptionsfaktor kann dann in den Zellkern der Zelle und aktivieren ein Reporter-Gen-System (in der Regel mit Ausdruck des HIS3, lacZ und ADE2 Gene), wodurch das Wachstum von Hefen auf selektiven Medien, die bezeichnend für Köder und Beute-Interaktion ist [ 2-4].
Die Identifizierung und Charakterisierung von integralen Membranproteinen Protein-Interaktionen werden Informationen zu helfen, verstehen ihre Funktion. Wie verstehen wir genauer und zerlegen die Rollen der Proteine, die mit integralen Membranproteinen interagieren, können wir einen Einblick in das dynamische Zusammenspiel der Regulierung dieser Proteine beteiligt zu gewinnen und zu entdecken neue Targets, dass therapeutisches Potential haben können.
2. Auswahl der Köder und geeignete MYTHOS-System
- Vor der Durchführung MYTHOS Analyse sicher, dass Ihr Protein seine N-und / oder C-Terminus hat im Cytosol der Zelle. Es ist wichtig, dass die C ub-LexA-VP16-Tag, um Ihre Protein in einem solchen Terminus fusioniert werden, da die deubiquitinating notwendigen Enzyme für Transkriptionsfaktor Freisetzung in das Cytosol befinden [4].
- Als nächstes entscheiden, welche der beiden großen Varianten des Mythos ist geeignet. Für nicht-native Hefe-Proteine, können traditionelle Mythos (tMYTH) verwendet werden, wobei Köder ektopisch sind aus einem Plasmid überexprimiert. Für native Hefe-Proteine, integriert ist MYTHOS (iMYTH) die Methode der Wahl. In iMYTH Köder sind endogen mit dem C ub-LexA-VP16-Tag markiert, so dass sie unter der Kontrolle ihres nativen Promotor. Dies ist vorteilhaft, als der Wildtyp-Expression von Ködern zu beseitigen Probleme mit Proteinüberexpression, wie eine erhöhte Anzahl von Fehlalarmen [3] assoziiert hilft. Mit Ausnahme der ersten Köder Bau und die selektive Medien verwendet werden, sind beide Formen des Mythos aus in eine im Wesentlichen gleiche Weise durchgeführt. Aus Gründen der Übersichtlichkeit werden wir uns hauptsächlich auf die Verwendung von tMYTH in diesem Bericht konzentrieren, da dieser Variante kann im Prinzip mit Membranproteinen aus nahezu jeder Organismus eingesetzt werden und istdamit breiter anwendbar.
3. Bait Generierung und-Validierung
- Benötigte Medien und Lösungen
- Sterile ddH 2 O durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten vorbereitet.
- 3-Amino-1 ,2,4-triazol (3-AT)-Lösung als 1M Stammlösung in ddH vorbereitet 2 O. Sterilisieren, indem es durch einen 0,2 um Filter.
- YPAD Growth Medien, bestehend aus 1% w / v Hefeextrakt, 2% w / v Pepton, 2% w / v Glucose und 100 uM Adenin in ddH 2 O. vorbereitet Sterilisieren durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten.
- 10x Amino Acid / Nucleotidbase Mix: Die komplette Mischung enthält 1,0 mM Adenin, 1,8 mM Uracil, 1,0 mM Arginin, 1,0 mM Histidin, 2,3 mM Isoleucin, 7,6 mM Leucin, 1,6 mM Lysin, Methionin 10,1 mm, 3,0 mm Phenylalanin, 16,8 mm Threonin, Tryptophan 2,0 mM, 1,7 mM Tyrosin und 12,8 mM Valin, in ddH 2 O vorbereitet. Für drop-out weglassen die notwendige Aminosäure (n) und / oder Nukleotid-Base (n). Sterilisieren durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten.
- Synthetische Drop-Out (SD) Growth Medien, bestehend aus 0,67% w / v Hefestickstoffbase (ohne Aminosäuren aber mit Ammoniumsulfat), 2% w / v Glucose, 2% w / v Agar und 1x Amino Acid / Nucleotide Mix, vorbereitet in ddH 2 O. Bereiten Sie sowohl flüssige als auch feste (mit 2% Agar) SD-Leucin Medien. Auch Herstellung fester SD-Tryptophan-Leucin und SD-Tryptophan-Leucin-Adenin-Histidin Medien. Sterilisieren durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten. Gießen festen Medien in 100x15 mm Petrischalen.
- Synthetische Drop-Out (SD) Growth Medium mit 3-AT. Bereiten SD-Tryptophan-Leucin-Adenin-Histidin Medien wie beschrieben, aber mit 3-AT bei Konzentrationen von 25, 50, 75 und 100 mm. Fügen Sie die entsprechende Menge an 3-AT aus dem 1M sterile Stammlösung zu den Medien, nachdem es wurde autoklaviert und abgekühlt (aber noch nicht verfestigt). Gießen Sie in 100x15 mm Petrischalen.
- PEG / Lithiumacetat Mix, bestehend aus 40% w / v PEG-3350, 120 mM Lithium-Acetat und 167 pg / ml Lachssperma-DNA (Typ III Natriumsalz) in ddH 2 O. vorbereitet Zur Sicherstellung der Sterilität dieser Mischung herzustellen aus sterilem Wasser und Lösungen (zB autoklaviert 50% PEG-3350, autoklaviert 1M Lithium-Acetat und 2 mg / ml Lachssperma-DNA-Typ III Natriumsalz in sterile ddH 2 O).
- Enzyme und Reagenzien für die Durchführung von PCR.
- Gewerbliche Miniprep Kit.
- Soda-Kalk-Glas Perlen (0,5 mm).
- Kompetente Escherichia coli-Zellen für Plasmid-Vermehrung (zB DH5a, XL10 Gold) und Standard-Medien für bakterielle Vermehrung und Plasmid-Selektion.
- Spezielle Hefestämme, Plasmide und Primer wie im Protokoll beschrieben.
- Generierung von tMYTH Baits von Gap Repair
- Der Köder muss in einen geeigneten Vektor für die Kennzeichnung und die Expression geklont werden. Eine Vielzahl von tMYTH Vektoren werden derzeit für den Einsatz in Köder Bau zur Verfügung. Vektoren wie die pCMBV4, pAMBV4 und pTMBV4 erlauben den Aufbau von C-terminal markierten Köder (Bait-C ub-LexA-VP16) unter der Kontrolle der CYC1 (schwach), ADH1 (stark) und TEF1 (sehr stark) Promotoren bzw. . N-terminal markiert Köder (LexA-VP16-C ub-Köder) kann mit Hilfe von Vektoren wie pTLB-1 und PBT3-N, die unter der Kontrolle des TEF1 und CYC1 Promotoren bzw. werden. Der Vektor Wahl hängt von der Köder und muss empirisch ermittelt werden. In bestimmten Fällen höhere Köder Ausdruck ist notwendig, um Wechselwirkungen zu erkennen, während in anderen Fällen Köder Überexpression tatsächlich sein kann schädlich sein, was zu einer erhöhten Anzahl von Fehlalarmen.
- Restriktionsverdau der ausgewählten Plasmid auf der entsprechenden Einschränkung Website (s). Die Spaltung sollte nur in unmittelbarer Nähe des C ub-LexA-VP16-Tag (vor dem Tag für C-terminal-Tagging oder nachgeschaltete für N-terminal-Tagging). Zum Beispiel, wenn Sie die pAMBV4 Vektor ist SfiI eine ideale Wahl. Bewahren Sie die verdaute Plasmid bei -20 ° C bis zur Verwendung.
- Design-Primer für die Amplifikation und Klonierung des Gens von Interesse. Die 5 'Ende der Vorwärtsprimer müssen übereinstimmen etwa 35-40 Nukleotide stromaufwärts von der Restriktionsstelle, während das 3'-Ende der ersten 18-20 Nukleotiden aus der Ziel-Gen übereinstimmen muss. Das 5'-Ende des Reverse-Primer muss mit dem reversen Komplement von etwa 35-40 Nukleotide hinter dem Restriktionsstelle, mit dem 3 'Ende Matching der reversen Komplemente der letzten 18-20 Nukleotiden aus der Ziel-Gen (unter Auslassung der Stop-Codon wenn die C ub-LexA-VP16 Tag wird an der C-Terminus platziert). Je nachdem, ob N-oder C-terminalen Tagging durchgeführt wird, wählen Sie die 35-40 Nukleotide der Vorwärts-oder Rückwärts-Primer, so dass das Ziel-Gen in frame mit dem C ub-LexA-VP16-Tag wird geklont.
- Amplify das Gen von Interesse mittels PCR unter Verwendung der oben Primer. PCR-Parameter hängt von der speziellen Enzym und spezifische Primer verwendet abhängen.
- Transform das PCR-Produkt zusammen mit der verdauten Plasmid in eine geeignete Hefe Laborstamms die mit einem leu2 Mutation (z. B. BY4741). Ein Mythos Reporter Belastungen (zB THY.AP4 oder L40) können verwendet werden, ist aber nicht notwendig, da der Zweck der Hefe an dieser Stelle einfach als ein Umfeld, in dem die Kluft Reparatur homologe Rekombination kann auftreten dienen. Transformation sollte wie folgt durchgeführt werden:
- Impfen eine einzelne Kolonie von Ihnen ausgewählten Hefestamm in 5 ml sterile YPAD Medien beimpft und über Nacht bei 30 ° C unter ständigem Schütteln (200 rpm).
- Verdünnen Sie die Übernacht-Kultur in 50 ml frisches YPAD Medien zu einer OD600 von ca. 0,15 und Inkubieren bei 30 ° C unter Schütteln (200 rpm). Wachsen Sie für ca. 3-4 Stunden, bis eine OD600 von ~ 0,6 erreicht ist.
- Zentrifugieren Sie die Zellen bei 700xg für 5 Minuten und entfernen Sie den Überstand.
- Zellpellet in 25 mL sterile ddH 2 O und Zentrifuge bei 700xg für 5 Minuten.
- Entfernen Sie den Überstand und Zellpellet in 1 ml sterilem ddH 2 O.
- Geben Sie 100 ul der Zellen, 300 ul PEG / Lithium-Acetat-Mix und die verdaute Plasmid (50 fmol) und PCR-Produkt (250-500 fmol) zu einem Mikrozentrifugenröhrchen.
- Inkubation bei 30 ° C für 30 Minuten.
- Hitzeschock bei 42 ° C für 1 Stunde.
- Zentrifuge bei 3000xg für 5 min und entfernen Sie den Überstand.
- Zellpellet in 200 ul sterilem ddH 2 O und Platte das gesamte Volumen auf feste, SD-Leucin selektiven Medien. Wachsen bei 30 ° C für 2-4 Tage.
- Grow up eine einzige Kolonie der transformierten Stamm in 5 ml SD-Leucin flüssigen Medien bei 30 ° C über Nacht.
- Centrifuge Zellen bei 700xg für 5 Minuten und entfernen Sie den Überstand.
- Isolieren Köder Plasmid-DNA aus den Zellpellets mit jeder kommerziellen Miniprep Kit. Folgen Sie den Standard-Protokoll mit einer Modifikation. Um eine ausreichende Hefezelle Lyse zu gewährleisten, fügen Sie ein kleines Volumen von 0,5 mm Kalk-Natron-Glas Perlen, um das Pellet nach der ersten Resuspendierung und kräftig vortexen für 5 Minuten. Dann mit dem kommerziellen Protokoll wie gewohnt fort.
- Transform isolierten Hefe-DNA in eine kompetente E. coli-Stamm für Plasmid-Vermehrung (zB DH5a, XL10 Gold) mit einem Transformationseffizienz von mindestens 1x10 7 Zellen / ug DNA. Beachten Sie, dass Köder Plasmide für die Verwendung von Kanamycin ausgewählt werden kann.
- Ernte Plasmid-DNA aus den transformierten E. coli mit einem Standard-DNA-Isolierung Methode oder kommerziellen Kits.
- Die ordnungsgemäße Errichtung der Köder-Plasmid durch Sequenzierung.
- Transform der geprüften Köder in eine geeignete MYTHOS Reporter Belastungen (zB THY.AP4, L40) zu konstruieren. Die Hefe-Transformation-Protokoll gerade beschrieben kann verwendet werden, ersetzen den Köder-Plasmid-DNA an die Stelle der verdauten Plasmid-und PCR-Produkt.
- Bait Validation - Richtiger Lokalisierung
- Vor dem Gebrauch sind Köder Stämme analysiert, um sicherzustellen, dass sie richtig sind, um die Hefe Membran lokalisiert ist. Bei der Verwendung von iMYTH, wird diese Lokalisation speziell hängen von den Eigenschaften des markierten Köder. Für tMYTH, Köder Plasmide umfassen im Allgemeinen eine Signalsequenz (zB Matα) Leitung des exprimierten Proteins an die Plasmamembran. Lokalisierung wird anhand der Fluoreszenzmikroskopie. Aufnahme eines YFP-Molekül in den Köder-Tag-Sequenz (dh C ub-YFP-LexA-VP16) ist die einfachste und direkteste Weg, so dass die direkte Visualisierung von lebenden Zellen, und wird üblicherweise in iMYTH verwendet. Alternativ kann ein Standard-Immunofluoreszenz-Ansatz unter Verwendung eines Antikörpers gegen das LexA oder VP16 Komponenten des Tags verwendet werden.
- Bait Validation - N ub G / I Control Test
- Einmal richtige Lokalisation der Köder hergestellt wurde, ist es notwendig, sicherzustellen, dass die Köder nicht aktiviert den Reporter-System allein oder in Gegenwart von nicht-interagierenden Beute (dh sicherzustellen, dass die Köder nicht selbst aktivieren). Dies geschieht mit Hilfe der N ub G / I-Test, wo der Köder mit Interaktion (positiv) und nicht-interagierenden (negative) Kontrolle Beute verwandelt, und das Wachstum auf selektiven Medien beurteilt. Ein Köder muss auf selektiven Medien zu wachsen in Anwesenheit der positiven Kontrolle, und nicht in Gegenwart der negativen Kontrolle wachsen, um geeignet zu sein für den Einsatz in MYTHOS.
- Beginnen Sie mit der Transformation der Köder anstrengen, um mit 100-200 ng der Kontrolle zum Opfer Plasmid getestet werden. Die zuvor beschriebenen Hefe-Transformation-Protokoll kann verwendet werden, ersetzen SD-Leucin Medien anstelle von YPAD und mit SD-Tryptophan-Leucin Medien für die endgültige Beschichtung Schritt. Die folgende Kontrolle Beute Konstrukte werden häufig verwendet:
- POST1-N ub I (Positive Control)
- POST1-N ub G (Negative Control)
- pFUR4-N ub I (Positive Control)
- pFUR4-N ub G (Negative Control)
- OST1 ist eine Komponente der Oligosaccharyltransferase komplex und wird an das endoplasmatische Retikulum Membran [9] lokalisiert, während FUR4 eine Uracil-Permease ist und an der Plasmamembran lokalisiert [10]. Obwohl diese Proteine in der Regel sind unsere erste Wahl für den Einsatz als nicht-interagierenden Beute, wird ihre Eignung von Fall zu Fall variieren. In dem unwahrscheinlichen Fall, dass Ihr Köder vorhergesagt wird, sich ernsthaft mit diesen beiden Steuerelementen interagieren, werden alternative Beute müssen gewählt werden. Daran erinnern, dass N ub I der Wildtyp-Form des N-Terminus von Ubiquitin ist, spontan und interagiert mit C ub unabhängig von einer Interaktion zwischen den Proteinen, denen die C-und N-ub ub verschmolzen sind. So N ub I Beute darstellen positive Kontrollen, während die N ub G Beute (mit der Isoleucin 13 bis Glycine Mutation, die spontane Assoziation von N ub und C ub verhindert) als negative Kontrollen dienen.
- Resuspendieren einzelne Kolonien von jedem verwandelt Köder in 100 ul sterilem ddH 2 O.
- Serienmäßig verdünnen die resuspendierten Zellen in sterile ddH 2 O, um Verdünnungen von 1 / 10, 1 / 100 und 1 / 1000 zu produzieren.
- Spot 5 ul Volumen von unverwässert und verwässert Zellen auf SD-Tryptophan-Leucin und SD-Tryptophan-Leucin-Adenin-Histidin Medien mit und ohne 3-AT bei verschiedenen Konzentrationen. 3-AT wirkt als kompetitiver Inhibitor des HIS3-Reportergens, und dient dazu, die Stringenz der Selektion zu erhöhen. Es kann in manchen Fällen nützlich zur Hemmung der unspezifischen Wachstum schwach bis mäßig Selbst-Aktivierung Köder.
- Lassen Sie die Flecken trocknen lassen und dann inkubieren die Platten bei 30 ° C für 2-4 Tage.
- Alle Transformanten sollten auf der SD-Tryptophan-Leucin-Platten zu wachsen, was darauf hinweist, dass sie erfolgreich mit Beute Plasmid transformiert. Baits, die nicht selbst aktivieren wird auf SD-Tryptophan-Leucin-Adenin-Histidin Medien wachsen nur, wenn sie mit N ub verwandelt I Beute Konstrukte, und nicht mit N ub G Beute. Beachten Sie, was Konzentration von 3-AT ist in den Medien erforderlich (falls vorhanden), da diese müssen während der Screening eingesetzt werden.
4. Abschirmung
- Benötigte Medien und Lösungen
- Sterile ddH 2 O durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten vorbereitet.
- 0,9% NaCl-Lösung in ddH 2 O hergestellt und sterilisiert durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten.
- Natriumphosphatlösung bestehend aus 493 mm Natriumdihydrogenphosphat und 250 mM Natriumphosphat einbasigen in ddH 2 O. Sterilisieren durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten.
- X-Gal (5-Brom-4-Chlor-3-Indolyl-β-D-galactopyranosid) Lösung als 100 mg / ml Stammlösung in N, N-Dimethylformamid vorbereitet.
- 2xYPAD Growth Medien mit 2% w / v Hefeextrakt, 4% w / v Pepton, 4% w / v Glucose und 100 uM Adenin, in ddH 2 O. vorbereitet Sterilisieren durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten.
- Synthetic Dropout (SD) Growth Media, wie zuvor beschrieben. Bereiten Sie flüssigem SD-Leucin und solide SD-Tryptophan-Leucin. Gießen Sie den festen Medien in beide 100x15 mm Petrischalen. Herstellung fester SD-Tryptophan-Leucin-Adenin-Histidin Medien in 150 mm Ronden, 16 Tafeln für jeden Bildschirm, mit 3-AT, falls erforderlich, bei der Konzentration von N ub G / I-Regelung ermittelt.
- Synthetic Dropout (SD) Media + 5-Bromo-4-Chlor-3-Indoyl-β-D-galactopyranosid (X-Gal). Bereiten SD-Tryptophan-Leucin-Adenin-Histidin-haltigen Medien-Agar wie zuvor beschrieben. Nach dem Autoklavieren, abkühlen lassen, fügen Sie 3-AT (falls erforderlich), gefolgt von 1:10 Volumen of sterile Natriumphosphatlösung. Als Nächstes fügen X-Gal-Lösung auf eine Endkonzentration von 80 ug / mL. Gründlich mischen und gießen Sie in 150 mm Ronden.
- PEG / Lithium-Acetat-Lösung II mit 40% PEG-3350, 100 mM Lithium-Acetat, 1 mM EDTA und 10 mM Tris pH 7,5. Diese Lösung mit sterilem ddH 2 O und Lösungen (zB autoklaviert 50% PEG-3350, 1 M Lithium-Acetat, 100 mM Tris pH 7,5 und 500 mM EDTA pH 8,0).
- Lithium-Acetat / Tris EDTA-Lösung mit 110 mM Lithium-Acetat, 11 mM Tris pH 7,5 und 1,1 mM EDTA. Diese Lösung mit sterilem ddH 2 O und Lösungen (zB autoklaviert 1M Lithium-Acetat, 100 mM Tris pH 7,5 und 500 mM EDTA pH 8,0).
- 10x Tris EDTA-Lösung, bestehend aus 100 mM Tris pH 7,5 und 10 mM EDTA in ddH 2 O. vorbereitet Sterilisieren durch Autoklavieren bei 121 ° C, 15 psi für 30 Minuten.
- Einsträngige Carrier-DNA (ssDNA)-Lösung mit 2 mg / ml Lachssperma DNA Type III Natriumsalz, in sterile ddH 2 O. vorbereitet
- Gewerbliche Miniprep Kit.
- Soda-Kalk-Glas Perlen (0,5 mm).
- Kompetente Escherichia coli-Zellen für Plasmid-Vermehrung (zB DH5a, XL10 Gold) und Standard-Medien für bakterielle Vermehrung und Plasmid-Selektion.
- Spezielle Hefestämme und Plasmide, wie in der Anleitung beschriebenen
- Large Scale Transformation
- Impfen einer einzelnen Kolonie des Mythos Reporter-Stamm, Ihre Köder in 5 ml SD-Leucin Medien und inkubieren bei 30 ° C über Nacht unter Schütteln (200 rpm).
- Verdünnen Sie die Übernacht-Kultur in 200 ml SD-Leucin Medien zu einer OD600 = 0,15 und Inkubieren bei 30 ° C unter Schütteln (200 rpm). Wachsen, bis die OD600 = 0,6 bis 0,7 (ca. 4-5 Stunden).
- Kurz vor dem Ziel OD600 erreicht ist, Tauwetter ein Aliquot von ssDNA-Lösung. Kochen bei 100 ° C für 5 Minuten und dann kühl auf Eis. Wiederholen Sie einmal.
- Wenn das Ziel OD600 erreicht worden ist, ernten die Zellen über Zentrifugation bei 700xg für 5 Minuten (teilen die 200 mL Kultur zwischen 4x50 mL Schraubdeckel-Zentrifugenröhrchen).
- Wash jedes Pellet mit 30 ml sterilem ddH 2 O und kurz vortexen der Probe. Zentrifuge bei 700xg für 5 Minuten.
- Verwerfen Sie den Überstand und resuspendieren jedes Pellet in 1 ml Lithium-Acetat / Tris EDTA-Lösung. Transfer in ein steriles 1,5 ml Mikrozentrifugenröhrchen und Zentrifuge bei 700xg für 5 Minuten.
- Verwerfen Sie den Überstand und resuspendieren jedes Pellet in 600 ul der Lithium-Acetat / Tris EDTA-Lösung.
- Fügen Sie Folgendes zu 4x15 mL Schraubdeckel-Zentrifugenröhrchen:
- 2,5 ml PEG / Lithium-Acetat-Lösung II
- 600 ul resuspendierten Zellen
- 100 ul ssDNA-Lösung
- 7 &mgr; g der Beute Bibliothek DNA
- Bibliotheken, die Beute entweder am N-oder C-Terminus mit N ub G markiert und hergestellt aus einer Vielzahl von cDNA-oder genomischen Quellen, sind kommerziell erhältlich ( www.dualsystems.com ). Die spezifische Bibliothek verwendet werden, müssen auf einer Fall-zu-Fall-Basis, abhängig von der gewählten Köder und experimentelle Ziele bestimmt werden.
- Beide Röhrchen für 1 Minute, um sicherzustellen, Durchmischung und dann inkubieren in einem 30 ° C im Wasserbad für 45 Minuten. Mix kurz alle 15 Minuten.
- Add 160 ul Dimethylsulfoxid (DMSO) in jedes Röhrchen und mischen Sie sofort durch Umdrehen der Röhrchen.
- Inkubieren in einem 42 ° C im Wasserbad für 20 Minuten.
- Sammeln Transformanten durch Zentrifugation bei 700xg für 5 Minuten.
- Überstand verwerfen. Recover Transformanten durch Aufwirbelung von jedem Pellet in 3 ml 2xYPAD. Pool alle Proben in einem einzigen 50-ml-Schraubdeckel-Zentrifugenröhrchen.
- Inkubation bei 30 ° C für 90 Minuten die Erholung der Zellen.
- Zentrifuge bei 700xg für 5 Minuten und den Überstand verwerfen.
- Resuspendieren der Zellpellets in 4,9 ml steriler 0,9% NaCl.
- Mit 100 L resuspendierten Zellen vorbereiten 10-fache serielle Verdünnungen in steriler 0,9% NaCl von 10x bis 1000x.
- Tafel 100 ul der 100x und 1000x Verdünnungen auf SD-Tryptophan-Leucin Medien und Inkubieren bei 30 ° C für 2-3 Tage. Diese Platten dienen als Kontrolle und werden verwendet, um die Effizienz der Transformation zu berechnen.
- Ebenso teilen die verbleibenden 4,8 ml resuspendierten Zellen und Platte auf große (150 mm) SD-Tryptophan-Leucin-Adenin-Histidin-Platten,mit den erforderlichen Betrag von 3-AT, wie in der N ub G / I-Test bestimmt, und inkubieren Sie bei 30 ° C für 3-4 Tage.
- Resuspendieren einzelne Kolonien (die jeweils Zellen, die eine mögliche Interaktion Köder-Beute-Paar) in 100 ul 0,9% NaCl und Platte 5 ul Aliquots auf SD-Tryptophan-Leucin-Adenin-Histidin + X-Gal-Medien (einschließlich 3-AT, wenn erforderlich). Lassen Sie die für 2-4 Tage wachsen. Dieser Schritt dient als eine zweite Runde des selektiven Screening und hilft bei der Beseitigung von Fehlalarmen in der ersten Runde gewonnen. Nur Kolonien, die ein robustes Wachstum und einen blauen Farb-Display für die weitere Analyse ausgewählt.
- Prey DNA Isolierung und Sequenzierung
- Isolieren Plasmid-DNA aus dem blauen Hefekolonien mit einer Miniprep-Protokoll mit den Änderungen vorher beschrieben. Achten Sie darauf, erwachsen zu werden die Zellen in SD-Tryptophan Medien nur, um für die Beibehaltung der Beute wählen, aber nicht Köder, Plasmide. Für Bildschirme, die eine sehr große Anzahl von Treffern zu erzeugen, kann ein im Handel erhältliches High-Throughput-Miniprep Kit an dieser Stelle von Vorteil.
- Transform isolierten Hefe-Plasmid-DNA in eine kompetente E. coli-Stamm für Plasmid-Vermehrung (zB DH5a, XL10 Gold) mit einem Transformationseffizienz von mindestens 1x10 7 Zellen / ug DNA. Beachten Sie, dass Beute-Plasmide für die Verwendung von Ampicillin kann ausgewählt werden.
- Isolieren Plasmid-DNA aus den transformierten E. coli mit einem Standard-DNA-Isolierung Methode oder kommerziellen Kits. Erneut kann ein High-Throughput-Miniprep Kit nützlich sein, wenn die Sample-Nummer ist groß. Die Amplifikation der DNA in E. coli erhöht Plasmidausbeute und sorgt dafür, dass eine ausreichende Menge an DNA vorhanden sowohl für die Sequenzierung und weitere Analysen.
- Sequence die isolierten Plasmide mit einem Primer ergänzen Sequenz innerhalb der N ub G.
- Kompilieren und analysieren alle Sequenzdaten Ihre vorläufige Liste der Interaktoren montieren. Dies kann manuell erfolgen oder in automatisierter Weise mit Hilfe geeigneter Software.
- Bait Dependency Testing
- Nach der Montage der vorläufigen Liste der Interaktoren ist es wichtig, überprüfen Sie die Wechselwirkungen und zu beseitigen promiscuous Beute, die / Interaktion aktivieren Sie die Reporter-Systems in einer Weise, unabhängig von Köder Identität. Dies geschieht mit Hilfe der Bait Dependency Test. In diesem Test werden alle der identifizierten Interaktionspartner wieder in den ursprünglichen Köder Belastung, sowie ein Stamm, eine Steuerung künstliche Köder, bestehend aus einer einzigen Transmembrandomäne fusioniert mit dem C ub-LexA-VP16-Tag umgewandelt. Transformation ist nach dem Standard-Protokoll zuvor beschrieben durchgeführt, wobei SD-Leucin Medien anstelle von YPAD und SD-Tryptophan-Leucin festen Medien für die endgültige Beschichtung Schritt.
- Resuspendieren einzelne Kolonien aus den obigen Transformationen in 100 ul sterilem ddH 2 O und Spot 5 ul auf SD-Tryptophan-Leucin-Adenin-Histidin + X-Gal-Medien (einschließlich 3-AT falls erforderlich). Die Platten werden dann für 2-4 Tage bei 30 ° C inkubiert Im Idealfall mehrere Transformanten sollte für jede Beute ausgewählt werden, und sowohl die ursprüngliche als Köder und Kunstköder sollten auf der gleichen Platte entdeckt werden.
- Hefe Durchführung der künstlichen Köder und Beute, die Ursache Aktivierung des Reporter-Systems (dh Wachstum und blaue Farbe) betrachtet werden promiscuous und dass bestimmte Beute aus der Liste der Interaktoren entfernt.
- Preys, dass Wachstum und Blaufärbung in Hefe Sache mit dem Köder-of-interest, aber nicht die künstliche Köder, bestätigt diese spezifische Wechselwirkung. Wenn jedoch, Beherbergung Hefe die Beute und Ihre Köder-of-interest nicht wachsen, ist diese Beute aus der Liste der Interaktoren entfernt.
- Die restlichen Beute bilden die komplette Liste der Interaktoren im Mythos Screen identifiziert werden.
5. Weitere Studien
Sobald MYTHOS Screening abgeschlossen ist, müssen weitere Analysen durchgeführt, um zu validieren und zu bestimmen, die biologische Bedeutung der nachgewiesenen Wechselwirkungen sein. Die Studien durchgeführt werden wird von Fall zu Fall variieren und müssen von den einzelnen Forscher bestimmt werden. Einige typische Beispiele für die Nachbereitung gehören Co-Immunopräzipitation Experimenten und Löschung Studien im nativen Organismus. Darüber hinaus können Computer-Analyse der erhaltenen Daten nützlich sein, zur Aufdeckung von Zusammenhängen, und helfen, die mögliche Relevanz und Rolle verschiedener Wechselwirkungen spielen zu identifizieren. Somit dient die MYTHOS-Technologie als einen sehr guten ersten Schritt "zur Identifikation und Verständnis für die kritischen funktionalen Wechselwirkungen von Membranproteinen. Zusammen mit detaillierten Follow-up-Studien und andere neu entwickelten und aufstrebenden technologies, verspricht es ein wertvolles Hilfsmittel bei der Erschließung der Geheimnisse der Zelle sein.

Abbildung 1. Das Prinzip der geteilten Ubiquitin. A. Die C-terminale Hälfte (C ub) und der N-terminalen Hälfte (N ub I): Ubiquitin kann in zwei Hälften getrennt werden. Diese Einheiten spontan rekonstruieren wegen ihrer hohen Affinität zu einem anderen. b. AN ub I Punktmutation an Position 13 aus einem Isoleucin zu Glycin (N ub G) verhindert, dass diese spontane Re-Vereins. c. Im Mythos-System wird die C ub, um den Köder-of-Interest (B) fusioniert und die Beute ist, die N ub G (A) fusioniert. AB Protein-Interaktion rekonstruiert pseudo-Ubiquitin.
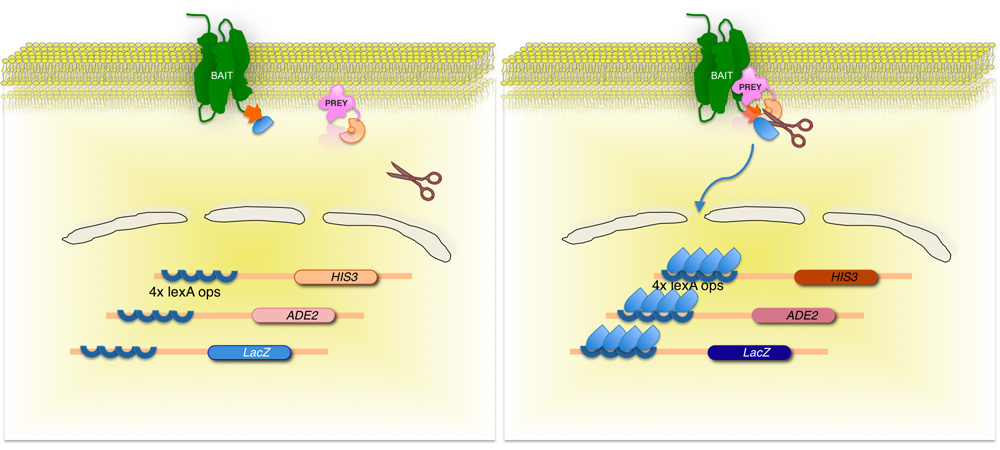
Abbildung 2. Split-Ubiquitin-basierten Membran Hefe-Zwei-Hybrid (Mythos) System.
Die Membran-Protein-of-Interest (Köder) ist es, die C-terminale Hälfte von Hefe-Ubiquitin (Ub C) fusioniert, konjugiert an einen Transkriptionsfaktor. Mit Hilfe eines cDNA oder gDNA Bibliothek ist jedes Protein durch die Bibliothek (Beute) codiert, um die entsprechenden N-Terminus des Ubiquitin-Einheit (N Ub G) fusioniert. Wenn die beiden Proteine nicht interagieren, bleibt der Transkriptionsfaktor an der Membran-Schnittstelle (links). Allerdings, wenn die Proteine interagieren, verbinden Sie die beiden Ubiquitin-Einheiten, die sich in der Spaltung durch Ubiquitin spezifischen Proteasen. Spaltung gibt den Transkriptionsfaktor, was Ausdruck der Reportergene (rechtes Bild)

Abbildung 3. MYTHOS Pipeline.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
MYTHOS ist das erste System mit hohem Durchsatz, die die Identifizierung von Wechselwirkungen zwischen full-length Membranproteinen und cytosolische oder membrangebundene Partnern ermöglicht. Es wurde verwendet, um Membranprotein aus einer Reihe von Organismen [3-7] zu studieren. Es gibt jedoch bestimmte Details, die müssen unter die Lupe, um sicherzustellen, das Protein-of-interest zugänglich ist mit MYTHOS Studie werden können.
Viele membrangebundene Proteine sind an die Plasmamembran über eine Signalsequenz, die anschließend gespalten wird, um das reife Protein produzieren gerichtet. Diese Sequenz ist keimspezifische und es ist möglich, dass diese native Signalsequenz wird mis-Lokalisation des Proteins-of-interest verursachen, wenn in Hefe exprimierten, weil die Signalsequenz bleibt unerkannt. Zur Umgehung dieses Problems, wir diese spezifische Proteine auf der Hefe-Signalsequenz, der Mating-Faktor-alpha (MATα) abgeleitet fusioniert werden, entwickelt. Dieses Peptid-Sequenz (sog. MFa-ss) re-lokalisiert das Protein der Hefe Plasmamembran und wichtiger ist, durch Hefe Signal-Peptidasen abgespalten. Diese Peptidsequenz in Plasmide pTMBV-MFa und pAMBV-MFa gefunden.
Ein weiterer wichtiger Parameter, der betont werden muss, ist Köder Expression. Der Veranstalter, dass die Expression des Köders Laufwerke regelt dieser Parameter. Es kann erforderlich sein Köder Expression mit dem NubG / Nubi-Test und die Höhe der zu optimieren 3-AT notwendig Überexpression Artefakte, wo der Köder ist "self-Aktivierung" (dh es wahllos interagiert mit vielen unspezifischen Beute Proteine) zu eliminieren . Untersuchen Hefe-Proteine produziert die physiologisch relevanten Konzentrationen Köder, wenn iMYTH angewendet wird. In diesem Fall ist das Gen-of-Interest mit der Cub-TF innerhalb der gDNA getaggt. Alternativ können exogene Proteine aus PBT3-STE und pCMBV Plasmide, dass die CYC1-Promotor was zu niedrigen Köder Ausdruck tragen ausgedrückt werden. Die Plasmide pTMBV und pTLB1 Hafen der TEF1-Promotor, während pAMBV die ADH1 Promotor hat, sowohl das Laufwerk starke Expression des Köderprotein. Wenn Köderprotein Ebenen weiter erfordern die Optimierung, kann es notwendig sein, um die pTLB-1 Plasmid, das die TEF1-Promotor trägt zu verwenden, jedoch ist die LexA DNA-bindende Domäne an R156G die Affinität zu den exogenen Reporter-Gen-Promotoren abklingen mutierte letztlich die Verringerung der Wahrscheinlichkeit der Selbst-Aktivierung [5].
Ein weiterer Faktor, eine wichtige Rolle spielt für MYTHOS Erfolg ist die Bibliothek der Wahl für das Screening-Verfahren eingesetzt. Dies wird auf der endogenen Köder Expressionsprofile ab. Zum Beispiel kann der Köder in spezifischen Geweben exprimiert werden, und deshalb ist es wichtig, eine Bibliothek, die aus diesem spezifischen Gewebe aufgebaut ist verwenden. Dadurch wird sichergestellt, physiologisch relevanten Wechselwirkungen erkannt werden.
Der Mythos ist eine einfache und schnelle Werkzeug, das eine Fülle von Informationen über eine Klasse von Proteinen, die schwer zu untersuchen sind zur Verfügung stellt. Diese identifizierten Wechselwirkungen können in der Aufklärung der volle biologische Funktion von Membranproteinen zu unterstützen.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Igor Stagljar ist ein Mitbegründer der Dualsystems Biotech, Schweiz.
Acknowledgments
Wir möchten Morgenröte Edmonds für eine kritische Lektüre des Manuskripts danken. Die Stagljar Labor wird aus Mitteln der kanadischen Stiftung für Innovation (CFI), die Canadian Institute for Health Research (CIHR), der Heart and Stroke Foundation, die Canadian Cancer Society und Novartis unterstützt.
Materials
| Name | Company | Catalog Number | Comments |
| Polyethenlene Glycol (PEG3350) | BioShop Canada | PEG335 | |
| Lithium Acetate Bihydrate | BioShop Canada | LIA001 | |
| X-Gal (5-Bromo-4-Chloro-3-Indolyl-b-D-galactopyranoside) | BioShop Canada | XGA001 | |
| N`,N-dimethyl formamide | BioShop Canada | DMF 451 | |
| 3-amino-1,2,4-triazole (3-AT) | BioShop Canada | ATT124 | |
| Sodium phosphate dibasic | BioShop Canada | SPD307 | |
| Sodium phosphate monobasic | Fisher Scientific | BP329-500 | |
| Salmon Sperm DNA | VWR international | CA80601-120 | |
| D-Glucose | BioShop Canada | GLU501 | |
| LB Broth LENOX | BioShop Canada | LBL405 | |
| Yeast Nitrogen Base | BioShop Canada | YNB406 | |
| Yeast Extract | BioShop Canada | YEX401 | |
| Peptone | BD Biosciences | 211677 | |
| Bio-Tryptone | BioShop Canada | TRP402 | |
| Adenine Sulphate | BioShop Canada | ADS201 | |
| L-Uracil | BioShop Canada | URA241 | |
| L-Threonine | BioShop Canada | THR002 | |
| L-Histidine | BioShop Canada | HIS200 | |
| L-Methionine | BioShop Canada | MET222 | |
| L-Valine | BioShop Canada | VAL201 | |
| L-Phenylalanine | BioShop Canada | PHA302 | |
| L-Isoleucine | BioShop Canada | ISO910 | |
| L-Tyrosine | BioShop Canada | TYR333 | |
| L-Leucine | BioShop Canada | LEU222 | |
| L-Arginine | BioShop Canada | ARG006 | |
| L-Tryptophane | Fisher Scientific | BP395-100 | |
| L-Lysine | BioShop Canada | LYS101 | |
| L-Alanine | Fisher Scientific | BP369-100 | |
| Agar | BioShop Canada | AGR001 | |
| Soda Lime Galss Beads | Biospec Products | 11079105 | |
| Sodium Chloride | BioShop Canada | SLD002 |
References
- Stagljar, I., Fields, S. Analysis of membrane protein interactions using yeast-based technologies. Trends Biochem Sci. 27 (11), 559-563 (2002).
- Iyer, K. Utilizing the split-ubiquitin membrane yeast two-hybrid system to identify protein-protein interactions of integral membrane proteins. Sci STKE. 275, pl3-pl3 (2005).
- Paumi, C. M. Mapping protein-protein interactions for the yeast ABC transporter Ycf1p by integrated split-ubiquitin membrane yeast two-hybrid analysis. Mol Cell. 26 (1), 15-25 (2007).
- Stagljar, I. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc Natl Acad Sci U S A. 95 (9), 5187-5192 (1998).
- Gisler, S. M. Monitoring protein-protein interactions between the mammalian integral membrane transporters and PDZ-interacting partners using a modified split-ubiquitin membrane yeast two-hybrid system. Mol Cell Proteomics. 7 (7), 1362-1377 (2008).
- Scheper, W. Coordination of N-glycosylation and protein translocation across the endoplasmic reticulum membrane by Sss1 protein. J Biol Chem. 278 (39), 37998-38003 (2003).
- Thaminy, S. Identification of novel ErbB3-interacting factors using the split-ubiquitin membrane yeast two-hybrid system. Genome Res. 13 (7), 1744-1753 (2003).
- Johnsson, N., Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A. 91 (22), 10340-10344 (1994).
- Kelleher, D. J., Gilmore, R. The Saccharomyces cerevisiae oligosaccharyltransferase is a protein complex composed of Wbp1p, Swp1p, and four additional polypeptides. J Biol Chem. 269 (17), 12908-12917 (1994).
- Chevallier, M. R. Cloning and transcriptional control of a eucaryotic permease gene. Mol Cell Biol. 2 (8), 977-984 (1982).
