Source: Michael S. Lee1 and Tonya J. Webb1
1 Department of Microbiology and Immunology, University of Maryland School of Medicine and the Marlene and Stewart Greenebaum Comprehensive Cancer Center, Baltimore, Maryland 21201
Immunohistochemistry (IHC) and immunocytochemistry (ICC) are techniques used to visualize the expression and localization of specific antigens using antibodies. The first published use of IHC was in 1941 when Albert Coons used the technique to visualize the presence of pneumococcal antigen in tissue sections from mice infected with Pneumococcus (1). The name, immunohistochemistry, is derived from the roots "immuno-," in reference to antibodies, and "histo-," in reference to the tissue sections used in IHC. The root "cyto-" in immunocytochemistry highlights the key difference between ICC and IHC. Whereas IHC uses sections of whole tissue, ICC uses cells that have been isolated from tissue or grown in culture. The difference in samples used means sample preparation technically differs between IHC and ICC, but otherwise the protocols for ICC and IHC are identical and one will find the terms are frequently used interchangeably.
In both IHC and ICC, antibodies with either chemical or fluorescent tags, such as peroxidase or rhodamine, respectively, are used to visualize the distribution of any antigen of interest through specific binding of the tagged antibody to the antigen. In the case of IHC, thin slices of tissue are immobilized on a slide to maintain the structure of the tissue before being stained, allowing the visualization of antigens in the context of whole tissues (Figure 1). In the case of ICC, cells are distributed evenly on a slide before being stained, allowing the visualization of antigen distribution within individual cells but not within the structure of any specific tissue. Due to the similarities between the two protocols, this protocol will focus on IHC to address the additional complexities of sample preparation involved in IHC.
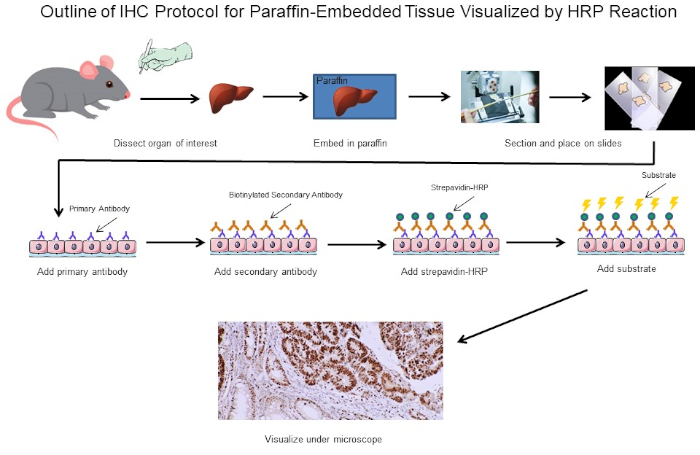
Figure 1: Outline of IHC Protocol. Visual outline of an IHC protocol for paraffin-embedded tissue dissected from a mouse. This protocol uses a biotinylated secondary antibody and strepavidin-HRP to visualize the location of antibody binding. Other options, such as fluorescently tagged antibodies, are also possible. Please click here to view a larger version of this figure.
The first major decision when performing IHC is how to prepare the tissue sections in order to maintain the structure of the tissue throughout the staining process. The two main choices are formalin-fixed sections of paraffin-embedded tissue or fresh sections of frozen tissue. There is no simple answer as to which method to use as it depends on what downstream analysis will be conducted. Formalin-fixation of paraffin embedded tissues is generally thought to better preserve tissue morphology for optimal imaging while freezing fresh tissue can preserve protein function for subsequent assays outside of IHC. In addition, fresh frozen tissue sections have been shown to be more suitable for gene expression analysis (2). A third consideration is whether or not the antibodies for your antigen of interest are suitable for either fixed or frozen tissue sections, as some antibodies have only been optimized for a specific type of section and may not work for others. Finally, one also needs to determine how long they need to store the tissue sections, as fresh frozen samples must be kept at -80°C and may not last beyond one year while fixed sections can be stored for much longer at room temperature. These are a few of the major considerations for determining whether to use formalin-fixed sections of paraffin-embedded tissue or fresh sections of frozen tissue. Ultimately, if one has enough tissue, it may be best just to have some of both.
In this experiment, we set out to determine if cyclin D1 expression was increased in enlarged spleens from a spontaneous mouse model of lymphoma development. Splenic tissue samples were first isolated from either wild-type mice, transgenic mice that do not have lymphoma, or transgenic mice that have spontaneously developed lymphoma. The spleen tissue samples were fixed in paraformaldehyde, embedded in paraffin, sectioned, stained using a mouse anti-cyclin D1 primary antibody followed by a horse anti-mouse secondary antibody, and developed using 3,3-diaminobenzidine (DAB). The sections were then counterstained in Harris Hematoxylin Solution and then the sections were imaged at 20X magnification.
Reagents
Paraffin-Embedded Sections
- 4% Paraformaldehyde (PFA)
- Ethanol (anhydrous denatured, histological grade 100%, 95%, 80%, 75%, and 50%). Can be diluted from 100% stock using double distilled water (ddH2O)
- Xylene
- IHC compatible glass slide to ensure tissue section remains attached throughout the entire procedure. IHC compatible glass slides have a specialized coating and are readily available from multiple retailers. If performing ICC, use a chambered slide. Chambered slides allow for cells to be seeded in the chambers and placed in the incubator until cells attach to the slide and reach proper confluency, at which point the chambers can be removed and staining can proceed in the same manner as IHC.
- Paraffin
- 0.3% Hydrogen peroxide (H2O2)/methanol: To prepare, add 1 mL 30% H2O2 to 99 mL methanol. Store at -20°C
- Antigen retrieval buffer: IHC citrate buffer pH 6.0
Fresh Frozen Sections
- Optimal cutting temperature (OCT) embedding compound
- Optimal fixative: 4% PFA or acetone that has been cooled to -20°C
Staining
- Blocking buffer: Should be determined by user. One example is horse serum diluted in 1X PBS
- Diluted primary antibody: see manufacturer specifications
- Diluted biotinylated secondary antibody: see manufacturer specifications
- Diluted avidin-Horseradish Peroxidase (HRP): Only for peroxidase visualization. See manufacturer specifications.
- DAB or another compatible substrate
- Counterstain (optional)
- Ethanol (anhydrous denatured, histological grade 100% and 95%)
- Xylene
- Organo/Limonene Mount
IHC and ICC have a vast range of applications. For example, one use of IHC is to examine the expression of oncogenes in spontaneous mouse models of tumor development. In Figure 2, we set out to determine if cyclin D1 expression was increased in enlarged spleens in a spontaneous mouse model of lymphoma development. Splenic tissue samples were fixed in paraformaldehyde, embedded in paraffin, sectioned, stained using an anti-cyclin D1 antibody (diluted 1:200 in blocking buffer), and then the sections were imaged at 20X magnification. Cyclin D1 expressing cells are indicated by the reddish-brown color against the blue tissue background. These results suggest that cyclin D1 expression was increased in enlarged spleens, indicating a correlation between cancer development and cyclin D1 expression in this model.

Figure 2: Splenic Cyclin D1 Expression in a Spontaneous Double Transgenic (DTG) Mouse Model of Lymphoma. An image of splenic tissue stained with an anti-Cyclin D1 primary antibody, counterstained with methyl green, and visualized using a biotinylated secondary antibody and ABC reagent activated with DAB substrate. The reddish-brown color represents locations where the antibody has bound indicating the presence of Cyclin D1 expressing tumor cells within the structure of splenic tissue that has been counterstained blue. Please click here to view a larger version of this figure.