1. Creation of a crude phage lysate (Figure 1)
- In a 50 ml flask, inoculate a fresh colony of LE392(λLZ1)[pPLate*D] (see Table 1 for details) into 6 ml of LB medium7 supplemented with 10 μg/ml Kanamycin and 100 μg/ml Ampicillin. Grow overnight at 30°C with mild shaking (180 rpm).
- Dilute the culture 1:100 into LBM (LB supplemented with 10 mM MgSO4) and grow at 30°C with mild shaking (180 rpm). In order to optimize the phage yield, make sure that the culture volume is no more than one-tenth of the flask volume capacity. We typically prepare two flasks of 2-liter or 2.5-liter capacity, and add 2.5 ml overnight culture into 250 ml LBM medium in each flask.
- When the cell density reaches OD600 ≈ 0.6 (~ 2.5 – 3 hr), induce the lysogen by moving the culture to a 42°C water bath shaker for 18 min with mild shaking (180 rpm), and then incubate at 37°C with mild shaking (180 rpm) until lysis is visible (culture becomes clear, in ~ 60 – 90 min).
- Add 2% chloroform to the culture, shake by hand to mix, and then incubate for 15 min at room temperature. Caution: Wear gloves to handle chloroform, and avoid breathing it.
- Transfer the culture into two 250 ml centrifuge bottles, centrifuge the culture in a Sorvall GSA rotor at 10,000 rpm for 15 min at 4°C. Recover the supernatant containing the phage particles, and discard the pellet of debris. Perform a second centrifugation to make sure to get rid of the visible debris.
- Use a standard phage titration protocol8 to measure the phage concentration. The phage titer should be ~ 5-10 x 109 pfu/ml. Use a supF strain such as LE392 as the indicator strain because of the Sam7 mutation in the genotype of the fluorescent phage, and use top agar and agar plates made with rich NZYM to obtain larger plaques (Figure 2).
2. Phage purification (Figure 1)
- Pour the lysate into a large (e.g. 2-liter) flask, add DNase I and RNase (1 μg/ml each) to the lysate in order to digest the nucleic acids liberated from lysed bacteria, and incubate 1 hr at room temperature.
- Add 1M NaCl to the lysate, transfer the lysate into 250 ml centrifuge bottles, and incubate 3 hr on ice. Centrifuge the lysate in a Sorvall GSA at 10,000 rpm for 15 min at 4°C. Recover the supernatant. The phage titer should be similar to that of the crude lysate, which is ~ 5-10 x 109 pfu/ml. The addition of NaCl promotes dissociation of phage particles from bacterial debris and is required for efficient precipitation of phage particles by PEG8.
- Pour the lysate into a large flask, e.g. 2-liter flask, add 10% (w/v) PEG8000 into the lysate, slowly stir or shake to dissolve PEG8000 at room temperature. Transfer the lysate into 250 ml centrifuge bottles and then incubate overnight (~ 16 hr) at 4°C. Centrifuge the lysate in a Sorvall GSA rotor at 10,000 rpm for 15 min at 4°C. Discard the supernatant.
- Soak the pellet (phage particles precipitated with PEG8000) with phage SM buffer (4 ml SM buffer per 250 ml of initial phage lysate). Incubate with very mild shaking or no shaking for 16 hr at 4°C.
- Gently take out the lysate (SM buffer with the phage particles) into a 50 ml Eppendorf centrifuge tube, and then wash the remaining pellet with 0.5 – 1 ml of SM buffer.
- Add equal volume of chloroform to the lysate. Gently mix the lysate with chloroform by inverting up and down for a few times. Centrifuge at 4,000 rpm for 15 min at 4°C in an Eppendorf 5804R or a similar bench-top centrifuge.
- Repeat Step 2.6 to get a clearer lysate. The phage titer should be ~ 1-2 x 1011 pfu/ml.
- Prepare SM/CsCl solutions with three different densities (ρ) of 1.3 g/ml, 1.5 g/ml and 1.7 g/ml. Measure the refractive index (η) to get a more accurate density reading. The density conversion9 is ρ = 10.8601 η – 13.4974 at 25°C. See Table 3 for details.
- Use a syringe with a long needle to load the solution into a 14 ml ultra-clear Beckman 40Ti ultracentrifuge tube. To avoid mixing and to form a better density gradient, underlaying the solution (i.e. layering solutions of increasing density under one another) should be used, i.e., gently load 2 ml of SM/CsCl solutions at the order of 1.3 g/ml, 1.5 g/ml and 1.7 g/ml by inserting the needle with a 3 ml syringe to the bottom of the tube.
- Gently load 8 ml of phage lysate by overlaying from the top of the 14 ml ultracentrifuge tube. Prepare a balance tube. Centrifuge in a Beckman SW40Ti rotor at 24,000 rpm for 4 hr at 4°C.
- Gently take out the tube in a dark room and illuminate from the top of the tube against a black background using a flashlight. The phage band should be clearly visible at the location of the interface between 1.3 g/ml and 1.5 g/ml SM/CsCl layers (Figure 3A). Puncture through the side of the tube slightly below the band with a 21.5 gauge needle with a 3 ml syringe. Gently collect ~ 500 μl of the phage suspension. The phage titer should be ~ 5-10 x 1011 pfu/ml.
- Place the phage suspension into a 4 ml ultra-clear Beckman ultracentrifuge SW60Ti rotor tube. Fill the tube with 1.5 g/ml SM/CsCl solution. Prepare a balance tube. Centrifuge in a Beckman SW60Ti rotor at 35,000 rpm for 24 hr at 4°C.
- Repeat the same procedure as in Step 2.11 to collect the phage from the visible band. The band should be visible as shown in Figure 3B.
- Load the phage suspension into a dialysis membrane cassette (Table 2) and dialyze three times against a 1000-fold volume of SM buffer at 4°C for durations of 3 hr, 3 hr and overnight (~ 16 hr). The purpose of the dialysis is to get rid of CsCl present in the phage suspension. The final phage titer should be ~ 5-10 x 1011 pfu/ml.
3. Prepare one agarose gel slab (Figure 4)
- Clean 6 microscope slides (75 x 50 mm, 1 mm thick) with 70% ethanol.
- Arrange 5 slides and secure with tape as shown in Figure 4.
- Mix 0.09 g agarose into 6 ml medium in a small beaker covered with cling wrap (yielding 1.5% agarose). Heat on a hot plate until the solution turns clear.
- Pour the agarose solution onto the secured slides.
- Place the last slide on top, carefully avoiding air bubbles. Place weight on top and allow to cool for ~ 30 min.
- Remove the 4 slides on the side, and wrap the slab together with the top and bottom slides with cling wrap. The slab can be stored at 4°C for up to 3 days.
4. Testing the purified phage stock
- Prepare a PBS-agarose gel slab as described above (Section 3).
- Stain the purified phage with DAPI. Mix 10 μl of phage (~ 1 x 1010 pfu/ml) with 10 μl of 10 μg/ml DAPI (final DAPI concentration of 5 μg/ml), incubate for 30 min at 4°C or 10 min at room temperature.
- Place 1 μl of the phage/DAPI mixture at the center of a No.1 24 x 50 mm coverslip, overlay a small piece (~ 10 x 10 mm) of the pre-prepared PBS-agarose slab. The small piece of agarose slab is cut with a razor blade after the top slide on the sandwich gel is removed. Image the sample under the epifluorescence microscope through the YFP and DAPI channels. Individual phages should be visible as diffraction-limited fluorescent “spots” in both channels (Figure 5). Use the same microscope and camera setups as in Step 6.2 below.
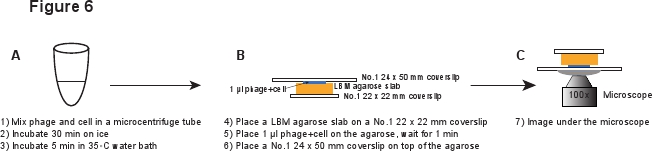
5. Infection (Figure 6)
- In a 14 ml Falcon tube, inoculate a fresh colony of LE392[pPRE-mCherry] (see Table 1 for details) into 2 ml of LB medium supplemented with 100 μg/ml Ampicillin, 10 mM MgSO4 and 0.2% Maltose. Grow overnight at 37°C with moderate shaking (265 rpm).
- Dilute the culture 1:1000 into LBMM (LB supplemented with 10 mM MgSO4 and 0.2% Maltose), i.e. add 5 μl overnight culture into 5 ml LBMM medium in a 50 ml flask. Grow to OD600 ≈ 0.4 at 37°C with moderate shaking (265 rpm).
- Use LBM medium to prepare an LBM-agarose gel slab as described in Section 3 above.
- Centrifuge 1 ml of cells at 2,000g in a bench-top microcentrifuge for 2 min at room temperature. Remove the supernatant, and gently resuspend the cells into 20 μl ice-cold LBMM to reach OD600 & 20.
- When manipulating the purified phage stock, use a wide pipette tip or cut the regular pipette tip to make the tip opening wider, to avoid shearing the phage particles3. Gently Mix 20 μl of cells with 20 μl of purified phage to reach an average phages-to-cell ratio in the range of 0.1 – 5. Incubate on ice for 30 min to allow phage adsorption, and then incubate in a 35°C water bath for 5 min to trigger phage DNA injection6.
- Pipette up and down a few times to separate any cell aggregates. Again use a wide pipette tip to avoid shearing the phages. Dilute the mixture 1:10 into LBMM, e.g. 5 μl mixture into 45 μl LBMM.
- Place a piece of LBM-agarose slab (~ 10 x 10 mm) on a No.1 22 x 22 mm coverslip. The pre-prepared LBM-agarose slab should be placed at room temperature for at least 1 hr before use to ensure that the agarose slab reaches room temperature. Place 1 μl of the phage/cell mixture on the agarose slab and wait for 1 min to allow the mixture to absorb into the agarose slab. Gently place a No.1 24 x 50 mm coverslip on the top of the agarose slab. This procedure is meant to avoid shearing the phages from the infected cell (Figure 6).
6. Following cell fate under the microscope
- Carefully mount the coverslip on the stage of the microscope. For imaging, use a high-magnification (e.g. 100x) objective (see Microscope System in Discussion below).
- Acquire an image set for the initial time frame. This image set will be used to characterize the initial numbers and positions of all infecting phages. Take a series of 15 images at 200 nm z-axis (vertical) intervals. Image through the YFP channel. In addition, take a single in-focus image through the phase-contrast and mCherry channels. Optimize the light intensity and exposure time to obtain sufficient signal while minimizing bleaching and cell damage (see Image Acquisition in Discussion below).
- Acquire a time-lapse movie of the post-infection cell fate. Image the sample in phase-contrast, YFP and mCherry channels at time intervals of 10 min for around 4 hr. During the time-lapse movie, use a single z-position image per channel per time point, to avoid unnecessary exposure of the sample, which could lead to bleaching and phototoxicity.
7. Image analysis
- Manually count the number of phages and record phage location and the cell length in the initial time frame. This can be done using software such as MetaMorph or ImageJ. Record the cell fates (lytic, lysogenic or uninfected), lysis time, and any other desired information by playing the time-lapse movie. To identify different cell fates, see Time-lapse Movie in Representative Results section below.
- In addition to the manual analysis above, more quantitative information (e.g. fluorescence level over time in individual cells) can be extracted by using automated cell-recognition and lineage tracing algorithms. We use a home built Matlab program for tracing the cell lineage and fluorescence levels, together with the Schnitzcell Matlab code for cell segmentation (written by the Elowitz group at Caltech).
8. Representative results:
Phage Plating:
The plaques of the fluorescently labeled phages (in Step 1.6 and Section 2) are significantly smaller than those of wild type (Figure 2). We therefore incubate the plates at least 12 hr in 37°C incubator for the plaques to be visible.
Phage Ultracentrifugation:
After ultracentrifugation of the phage sample with the CsCl step gradient (Step 2.10), two bands should be visible (Figure 3A). The top band, at the interface between the phage suspension and SM/CsCl 1.3 g/ml layer, contains cell debris and empty phage capsids. The bottom band, at the interface between SM/CsCl 1.3 g/ml and 1.5 g/ml layers, is the phage band. This band appears greenish for the fluorescent phage λLZ2. The band for wild type phage λIG2903 appears bluish5. After the ultracentrifugation of CsCl equilibrium gradient in Step 2.12, one phage band should be visible in the middle part of the tube (Figure 3B). Since the fluorescent phage λLZ2 contains a mixture of gpD-EYFP and gpD capsids, the ratio of protein-to-DNA is higher than that of wild type. Therefore, the band of the fluorescent phage λLZ2 is slightly lighter (appears to be at a higher location in the tube) than that of wild type λIG290310.
DAPI Staining:
Figure 5 shows typical images obtained after labeling the phage with DAPI (Section 4). The YFP and DAPI signals of a successfully purified phage should have close to 100% correspondence. We typically observe that less than 1% of the YFP spots do not contain DAPI (representing capsids without the viral genome). Less than 1% of the DAPI spots do not contain YFP (corresponding to non-fluorescent phages)5.
Time-lapse Movie:
Lytic cells are recognized by the accumulation of YFP fluorescence (green channel) inside the cell, followed by cell lysis. Lysogenic cells are recognized by the accumulation of uniform mCherry fluorescence (red) inside the cell and the resumption of normal cell growth and division. Uninfected cells (or cells where infection has failed) will not display any of the phenotypes above and will grow and divide normally. Figure 7 shows a few image-sets of phase-contrast, YFP and mCherry channels, and the corresponding overlaid images of these three channels, from a typical time-lapse movie (Section 6). The individual phages (green spots) are clearly visible at the initial time frame (Figure 7A). Typically, a number of phages are seen on the cell surface (presumably infecting those cells) while other phages are unadsorbed, as shown in Figure 7B (left panel). The infection outcome becomes distinguishable over time. The lytic cycle is indicated by the intracellular production of new phages (green, Figure 7C) followed by cell lysis (exploded cells with released green phages, Figure 7D). Lysogeny is indicated by the production of mCherry from the PRE promoter (red, Figure 7C) and the resumption of cell growth and division (red, Figure 7D).




Figure 1. Flow chart describing the creation of fluorescent phages. A crude phage lysate is first obtained by inducing a lysogen of the gpD-EYFP phage, harboring a plasmid expressing wild type gpD protein (panels A-B). The phage is purified through a series of steps (panels C-L).

Figure 2. Phage plaques. Plaques of the fluorescent phage (left) are smaller than those of wild type (right) after incubating plates for 12 hr at 37°C.

Figure 3. Phage bands after ultracentrifugation. A) Two bands are visible after ultracentrifugation in a CsCl step gradient. The top one corresponds to cell debris and empty phage capsids; the bottom band contains the desired phage. Left: fluorescent phage, right: wild type. B) A single phage band is visible after ultracentrifugation in a CsCl equilibrium gradient. The fluorescent phage band (left) is greenish, compared to a bluish band for wild type phage (right).

Figure 4. The procedure of preparing agarose gel slabs.

Figure 5. Fluorescent images of phages after DAPI staining. Individual phages are easily distinguishable, and YFP and DAPI signals co-localize very well.

Figure 6. Schematic description of phage infection and imaging setup. Click here to view a full-sized version of this image.

Figure 7. Typical images from a time-lapse movie of phage infection. Shown are the phase-contrast, YFP and mCherry channels, as well as an overlay of the three channels. (A) YFP-channel images from the initial time frame. Left, the sum of YFP images at different z-positions. The three right images are sample YFP images at different z-positions, corresponding to different areas of the cell surface. (B), (C) and (D) Overlaid images (left) of the phase-contrast (middle-left), YFP (middle-right) and mCherry (right) channels at different time frames. (B) At t = 0, two cells are seen, each infected by a single phage (green spots), and one cell is infected by 3 phages. Also observed are some unadsorbed phages. (C) At t = 80 min, the two cells infected by single phages have each gone into the lytic pathway, as indicated by the intracellular production of new phages (green). The cell infected by 3 phages has gone into the lysogenic pathway, as indicated by the production of mCherry from the PRE promoter (red). (D) At t = 2 hr, the lytic pathway has resulted in cell lysis (cell exploded), while the lysogenic cell has divided§.
§Left panels of Figure 7(C) and (D) are reprinted from Cell, 141, Lanying Zeng, Samuel O. Skinner, Chenghang Zong, Jean Sippy, Michael Feiss, and Ido Golding, Decision Making at a Subcellular Level Determines the Outcome of Bacteriophage Infection, 682-691, Copyright (2010), with permission from Elsevier.
| Strain name | Relevant genotype | Source/reference |
| Bacterial strains | ||
| LE392 | supF | John Cronan, University of Illinois |
| Phage strains | ||
| λLZ1 | gpD-EYFP, cI857 Sam7 D-eyfp b::kanR | Zeng et al.5 |
| λLZ2 | gpD-mosaic, same genotype as λLZ1 | Zeng et al.5 |
| Plasmids | ||
| pPRE–mCherry | mCherry under the control of PRE, ampR | Zeng et al.5 |
| pPLate*D | gpD under the control of λ late promoter, ampR | Zeng et al.5 |
Table 1. Bacterial strains, phages and plasmids used in this work.
| Density ρ (g/ml) | CsCl (g) | SM (ml) | Refractive Index η |
| 1.30 | 39 | 86 | 1.3625 |
| 1.50 | 67 | 82 | 1.3815 |
| 1.70 | 95 | 75 | 1.3990 |
Table 3. CsCl solutions prepared in SM buffer (100 ml) for step gradients.

{kind=link}