

Summary
In questo articolo viene descritta la procedura per la preparazione di una versione fluorescente marcato del batteriofago lambda, infezione di
Abstract
Il sistema comprendendo batteriofago (fago) lambda e del batterio E. coli si è servito a lungo come paradigma per la cella-destino 1,2 determinazione. Seguendo l'infezione simultanea della cella da un numero di fagi, uno dei due percorsi è scelto: litico (virulenta) o lisogena (dormiente) 3,4. Abbiamo recentemente sviluppato un metodo per l'etichettatura fluorescente fagi singoli, e sono stati in grado di esaminare la post-infezione decisionale in tempo reale al microscopio, a livello di fagi singoli e cellule 5. Qui, descriviamo la procedura completa per eseguire gli esperimenti di infezione descritti nel nostro precedente lavoro 5. Questo include la creazione di fagi fluorescenti, infezione delle cellule, l'imaging al microscopio e l'analisi dei dati. Il fago fluorescente è un "ibrido", co-esprimono versioni wild-type e YFP-fusione della proteina del capside GPD. Un lisato fago grezzo viene prima ottenuto inducendo una lysogen del GPD-EYFP (Enhbilanciato giallo fluorescente delle proteine) fago, ospitare un plasmide che esprime di tipo selvatico GPD. Una serie di fasi di purificazione vengono poi effettuate, seguito da DAPI-etichettatura e immagini al microscopio. Questo viene fatto al fine di verificare l'uniformità, confezionamento efficienza DNA, segnale di fluorescenza e la stabilità strutturale del magazzino fago. L'adsorbimento iniziale di fagi a batteri viene eseguita su ghiaccio, seguito da una breve incubazione a 35 ° C per attivare l'iniezione del DNA virale 6. Il fago / miscela di batteri viene poi spostato alla superficie di una lastra sottile agar nutriente, coperto con un vetrino coprioggetto e ripreso sotto un microscopio a epifluorescenza. Il post-infezione processo è seguito per 4 ore, a intervallo di 10 min. Posizioni a più stadi sono tracciati in modo tale che ~ 100 infezioni delle cellule può essere rintracciata in un singolo esperimento. Ad ogni punto di posizione e tempo, le immagini vengono acquisite in contrasto di fase e fluorescenti canali rosso e verde. Il contrasto di fase immagine è utilizzato in seguito per l'assemblaggio cell riconoscimento mentre i canali fluorescenti sono usate per caratterizzare il risultato dell'infezione: produzione di nuovi fagi fluorescenti (verde) seguita da lisi cellulare, o l'espressione di fattori lisogenia (rosso), seguito da crescita cellulare ripresa e divisione. Gli acquisiti filmati time-lapse sono trattati con una combinazione di metodi manuali e automatizzati. I risultati delle analisi dei dati di identificazione dei parametri di infezione per ogni evento infezione (ad esempio il numero e le posizioni di infettare fagi), così come risultato l'infezione (lisi / lisogenia). Ulteriori parametri possono essere estratti se desiderato.
Protocol
1. Creazione di un lisato fagico grezzo (figura 1)
- In un pallone da 50 ml, inoculare una colonia fresca LE392 (λ LZ1) [pPLate * D] (vedi Tabella 1 per i dettagli) in 6 ml di terreno LB 7 integrato con 10 ug / ml kanamicina e 100 pg / ml di ampicillina. Crescere durante la notte a 30 ° C, con agitazione lieve (180 rpm).
- Diluire il 1:100 cultura in LBM (LB supplementato con 10 mM MgSO4) e crescere a 30 ° C, con agitazione lieve (180 rpm). Per ottimizzare la resa fago, assicurarsi che il volume cultura non è più di un decimo della capacità di volume del flacone. Noi di solito preparare due fiaschi di capacità 2 litri o 2,5 litri, e aggiungere 2,5 ml di coltura durante la notte in 250 ml di terreno LBM in ogni contenitore.
- Quando la densità cellulare raggiunge OD 600 ≈ 0,6 (~ 2,5-3 ore), inducono la lysogen spostando la cultura ad un 42 ° C in bagno di acqua agitatore per 18 minuti con agitazione lieve (180 rpm), e poi incubatE a 37 ° C, con agitazione lieve (180 rpm) fino a lisi è visibile (la cultura diventa chiaro, in ~ 60 - 90 min).
- Aggiungere 2% cloroformio alla cultura, agitare manualmente per mescolare e incubare per 15 min a temperatura ambiente. Attenzione: Indossare guanti per gestire cloroformio, ed evitare di respirarlo.
- Trasferire la cultura in bottiglie ml due 250 centrifuga, centrifugare la cultura in un GSA Sorvall rotore a 10.000 rpm per 15 min a 4 ° C. Recuperare il surnatante contenente le particelle fagiche, e scartare il pellet di detriti. Eseguire una seconda centrifugazione per fare in modo di sbarazzarsi dei detriti visibile.
- Utilizzare un protocollo standard titolazione fago 8 per misurare la concentrazione di fago. Il titolo fago dovrebbe essere ~ 5-10 x 10 9 pfu / ml. Utilizzare un ceppo supF come LE392 come il ceppo indicatore a causa della mutazione SAM7 nel genotipo del fago fluorescente, e utilizzare agar top e piastre di agar fatto con NZYM ricchi per ottenere più grandi placche (Figura 2).
2. Fago purificazione (Figura 1)
- Versare il lisato in una grande (ad esempio 2 litri) matraccio DNasi e RNasi (1 mg / ml) al lisato per digerire gli acidi nucleici liberate da batteri lisati, e incubare per 1 ora a temperatura ambiente.
- Aggiungi 1M NaCl al lisato, trasferire il lisato in bottiglie da 250 ml centrifuga, e incubare 3 ore sul ghiaccio. Centrifugare il lisato in una Sorvall GSA a 10.000 rpm per 15 min a 4 ° C. Recuperare il surnatante. Il titolo fago dovrebbe essere simile a quella del lisato grezzo, che è ~ 5-10 x 10 9 pfu / ml. L'aggiunta di NaCl promuove la dissociazione di particelle fagiche da detriti batterici ed è necessario per precipitazione efficace di particelle fagiche da PEG 8.
- Versare il lisato in un pallone grande, ad esempio pallone da 2 litri, aggiungere 10% (w / v) PEG8000 nel lisato, lentamente mescolare o agitare per sciogliere PEG8000 a temperatura ambiente. Trasferire il lisato in 250 cent mlbottiglie rifuge e poi incubare una notte (~ 16 ore) a 4 ° C. Centrifugare il lisato in un rotore Sorvall GSA a 10.000 rpm per 15 min a 4 ° C. Eliminare il surnatante.
- Immergere il pellet (particelle fagiche precipitato con PEG8000) con fago tampone SM (4 ml tampone SM per 250 ml di lisato fagico iniziale). Incubare con agitazione molto lieve o nessuna agitazione per 16 ore a 4 ° C.
- Delicatamente estrarre il lisato (buffer di SM con le particelle fagiche) in una centrifuga tubo da 50 ml Eppendorf, e poi lavare il restante pellet con 0,5 - 1 ml di tampone SM.
- Aggiungere uguale volume di cloroformio al lisato. Mescolare delicatamente il lisato con cloroformio fino invertendo e giù per un paio di volte. Centrifugare a 4000 rpm per 15 min a 4 ° C in un 5804R Eppendorf o simile banco centrifuga.
- Ripetete il passo da 2,6 a ottenere un lisato più chiara. Il titolo fago dovrebbe essere ~ 1-2 x 10 11 pfu / ml.
- Preparare SM / CsCl soluzioni con tre diverse densità (ρ) di 1,3 g / ml, 10,5 g / ml e 1,7 g / ml. Misurare l'indice di rifrazione (η) per ottenere una lettura di densità più accurata. La conversione di densità 9 è ρ = η 10,8601 - 13,4974 a 25 ° C. Vedere la Tabella 3 per i dettagli.
- Utilizzare una siringa con un ago lungo per caricare la soluzione in un 14 ml ultra-trasparente tubo ultracentrifuga Beckman 40Ti. Per evitare la miscelazione e per formare un gradiente di densità maggiore, come sfondo per la soluzione (soluzioni stratificazione cioè densità in aumento sotto un altro) deve essere utilizzato, cioè, delicatamente caricare 2 ml di SM / CsCl soluzioni al fine di 1,3 g / ml, 1,5 g / ml e 1,7 g / ml inserendo l'ago di una siringa 3 ml al fondo della provetta.
- Delicatamente caricare 8 ml di lisato fagico sovrapponendo dalla sommità del tubo un'ultracentrifuga 14 ml. Preparare un tubo di equilibrio. Centrifugare in un rotore Beckman SW40Ti a 24.000 rpm per 4 ore a 4 ° C.
- Delicatamente estrarre il tubo in una stanza buia e illuminare dalla parte superiore del tubo contro uno sfondo nero usando aflashlight. La banda fago dovrebbe essere chiaramente visibile nella posizione dell'interfaccia tra 1,3 g / ml e 1,5 g / ml SM / CsCl strati (figura 3A). Perforare attraverso il lato del tubo leggermente inferiore alla banda con un ago calibro 21,5 con una siringa 3 ml. Delicatamente raccogliere ~ 500 microlitri della sospensione fago. Il titolo fago dovrebbe essere ~ 5-10 x 10 11 pfu / ml.
- Posizionare la sospensione fago in un ml 4 ultra-trasparente tubo ultracentrifuga Beckman SW60Ti rotore. Riempire la provetta con 1,5 g / ml SM / soluzione CsCl. Preparare un tubo di equilibrio. Centrifugare in un rotore Beckman SW60Ti a 35.000 rpm per 24 ore a 4 ° C.
- Ripetere la stessa procedura della Fase 2,11 per raccogliere il fago dalla banda visibile. La banda dovrebbe essere visibile come mostrato in figura 3B.
- Caricare la sospensione fago in una cassetta membrana di dialisi (Tabella 2) e tre volte dializza contro un 1.000 volte il volume di tampone SM a 4 ° C per una durata di 3 ore, 3 ore e overnight (~ 16 ore). Lo scopo della dialisi è di liberarsi di CsCl presenti nella sospensione fago. Il titolo fago finale dovrebbe essere ~ 5-10 x 10 11 pfu / ml.
3. Preparare una lastra di gel di agarosio (Figura 4)
- Pulizia 6 vetrini da microscopio (75 x 50 mm, 1 mm di spessore) con il 70% di etanolo.
- Disporre 5 vetrini e fissare con nastro come mostrato nella Figura 4.
- Mescolare 0,09 g di agarosio in 6 ml di mezzo in un piccolo becher coperto con pellicola trasparente (producendo 1,5% agarosio). Scaldare su una piastra calda fino a quando la soluzione diventa chiara.
- Versare la soluzione di agarosio sui fogli protetti.
- Posizionare l'ultima diapositiva in alto, evitando accuratamente di bolle d'aria. Collocare il peso sulla parte superiore e lasciarlo raffreddare per ~ 30 min.
- Rimuovere le 4 diapositive sul lato, e avvolgere la lastra con le diapositive superiore e inferiore con pellicola trasparente. La lastra può essere conservato a 4 ° C per 3 giorni.
4. Test del magazzino fago purificato
- Preparareun PBS-lastra gel agarosio come descritto in precedenza (sezione 3).
- Macchiare il fago purificato con DAPI. Miscelare 10 ml di fago (~ 1 x 10 10 pfu / ml) con 10 pl di 10 pg / ml DAPI (concentrazione finale DAPI di 5 mg / ml), incubare per 30 min a 4 ° C o 10 minuti a temperatura ambiente.
- Posto 1 microlitri della fago / DAPI miscela al centro di una No.1 24 x 50 mm coprioggetti, overlay un piccolo pezzo (~ 10 x 10 mm) del pre-preparata PBS-agarosio lastra. Il piccolo pezzo di lastra agarosio viene tagliata con una lama di rasoio dopo la slitta superiore del gel sandwich viene rimosso. Immagine il campione sotto il microscopio a epifluorescenza attraverso i canali YFP e DAPI. Fagi individuali dovrebbe essere visibile come diffrazione limitata fluorescenti "punti" in entrambi i canali (Figura 5). Usate il microscopio stesso e le impostazioni della fotocamera come indicato al punto 6.2.
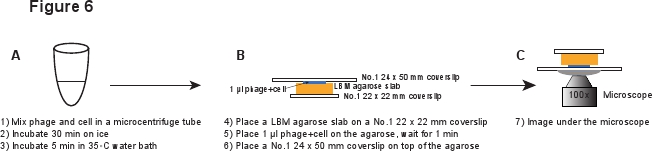
5. Infezione (Figura 6)
- In un tubo di 14 ml Falcon, inoculare una colonia fresca di LE392 [pP RE-MCHerry] (vedi Tabella 1 per i dettagli) in 2 ml di terreno LB supplementato con 100 pg / ml di ampicillina, 10 mM MgSO 4 e 0,2% maltosio. Crescere durante la notte a 37 ° C, con agitazione moderata (265 rpm).
- Diluire il 1:1000 cultura in LBMM (LB supplementato con 10 mM MgSO4 e 0,2% maltosio), vale a dire aggiungere 5 microlitri cultura durante la notte in 5 ml di terreno LBMM in un pallone da 50 ml. Crescere per OD 600 ≈ 0,4 a 37 ° C, con agitazione moderata (265 rpm).
- Utilizzare medio LBM per preparare un LBM-agarosio lastra gel come descritto nella sezione 3 di cui sopra.
- Centrifugare 1 ml di cellule a 2.000 g in una microcentrifuga da banco per 2 min a temperatura ambiente. Rimuovere il surnatante e sospendere nuovamente le cellule delicatamente in 20 microlitri ghiacciata LBMM raggiungere OD 600 e 20.
- Quando la manipolazione dello stock fago purificato, utilizzare una punta larga pipetta o tagliare la punta della pipetta regolare per rendere la punta di apertura più ampio, per evitare cesoiamento le particelle fagiche 3. Delicatamente Mix 20 microlitri di cellule con 20 pl di fago purificato per raggiungere una media fagi-cellula rapporto nel range di 0,1 - 5. Incubare su ghiaccio per 30 min per permettere fago adsorbimento, e poi incubare a 35 ° C in bagno d'acqua per 5 minuti per attivare l'iniezione del DNA fagico 6.
- Pipettare su e giù alcune volte per separare eventuali aggregati cellulari. Anche in questo caso utilizzare una punta larga pipetta per evitare cesoiamento i fagi. Diluire il composto in LBMM 01:10, per esempio 5 miscela in 45 microlitri LBMM pl.
- Posizionare un pezzo di LBM-agarosio lastra (~ 10 x 10 mm) su una No.1 coprioggetto 22 x 22 mm. Il pre-preparato LBM-agarosio lastra deve essere posto a temperatura ambiente per almeno 1 ora prima dell'uso per assicurare che la lastra di agarosio raggiunge la temperatura ambiente. Posto 1 microlitri della fago / cella miscela sulla lastra di agarosio ed attendere 1 min per permettere alla miscela di assorbire nella lastra agarosio. Cautela, inserire un No.1 coprioggetto 24 x 50 mm in alto della lastra agarosio. Questa procedura ha lo scopo di evitare il taglio dei fagi dal infcella perturbato (Figura 6).
6. In seguito il destino delle cellule sotto il microscopio
- Con cautela il coprioggetto sul palco del microscopio. Per l'imaging, utilizzare un obiettivo ad alto ingrandimento (es. 100x) (vedi Sistema Microscopio in discussione sotto).
- Acquisire un'immagine fissata per il periodo di tempo iniziale. Questo insieme immagine sarà utilizzata per caratterizzare i numeri iniziali e le posizioni di tutti i fagi infettano. Prendete una serie di 15 immagini a 200 nm asse z (verticale) intervalli. Immagine attraverso il canale YFP. Inoltre, prendere un singolo di messa a fuoco l'immagine attraverso il contrasto di fase e canali mCherry. Ottimizzare l'intensità della luce e il tempo di esposizione per ottenere un segnale sufficientemente riducendo al minimo sbianca e danno cellulare (vedere Acquisizione di immagini in discussione sotto).
- Acquisire un time-lapse film del post-infezione destino cellulare. Immagine del campione in contrasto di fase, YFP e mCherrycanali a intervalli di tempo di 10 minuti per circa 4 ore. Durante il film accelerato, utilizzare un singolo z-posizione dell'immagine per canale per punto di tempo, per evitare l'esposizione non necessaria del campione, che potrebbe portare a candeggio e fototossicità.
7. Analisi delle immagini
- Manualmente contare il numero di fagi e fago posizione di registrazione e la lunghezza della cella nel lasso di tempo iniziale. Questo può essere fatto utilizzando software come MetaMorph o ImageJ. Registrare i destini cellulari (litico, lisogenico o non infetto), il tempo di lisi, e tutte le altre desiderato suonare il time-lapse film. Per identificare destini cellulari diversi, vedere time-lapse video nella sezione rappresentativa risultati qui sotto.
- In aggiunta alle analisi manuale di cui sopra, ulteriori informazioni quantitative (ad esempio, livello di fluorescenza nel tempo in singole cellule) può essere estratta con automatizzate algoritmi di riconoscimento cellulare e lineage tracing. Usiamo un programma costruito casa Matlab per tracing linea cellulare e livelli di fluorescenza, insieme al codice Schnitzcell Matlab segmentazione per cella (scritto dal gruppo Elowitz Caltech).
8. Rappresentante dei risultati:
Phage Placcatura:
Le placchette dei fagi contrassegnati in modo fluorescente (in 1,6 Fase e sezione 2) sono significativamente inferiori a quelli di tipo selvatico (Figura 2). Abbiamo quindi incubare le piastre di almeno 12 ore in incubatore a 37 ° C per le placche siano visibili.
Phage ultracentrifugazione:
Dopo ultracentrifugazione del campione fago con il gradiente di CsCl passo (Passo 2,10), due bande devono essere visibili (Figura 3A). La banda superiore, all'interfaccia tra la sospensione fagica e SM / CsCl 1,3 g / ml strato, contiene detriti cellulari e capsidi fagiche vuote. La banda inferiore, all'interfaccia tra SM / CsCl 1,3 g / ml e 1,5 g / ml strati, è la banda fago. Thibanda s appare verdastro per la fluorescente fago λ LZ2. La band di tipo selvatico fago λ IG2903 appare bluastro 5. Dopo l'ultracentrifugazione su gradiente di CsCl equilibrio nella Fase 2,12, una banda fago dovrebbe essere visibile nella parte centrale del tubo (Figura 3B). Poiché la fluorescenza fago λ LZ2 contiene una miscela di GPD-EYFP e GPD capsidi, il rapporto di proteina-DNA è superiore a quella del tipo selvatico. Pertanto, la banda del fago λ fluorescente LZ2 è leggermente più leggero (sembra essere in una posizione più alta nel tubo) rispetto a quella di tipo selvatico λ IG290310.
DAPI colorazione:
La Figura 5 mostra immagini tipiche ottenuti dopo l'etichettatura del fago con DAPI (sezione 4). I segnali YFP e DAPI di un fago successo purificato deve avere quasi il 100% corrispondenza. Abbiamo tipicamente osservare che meno dell'1% della macchia YFPs non contengono DAPI (che rappresenta capsidi senza il genoma virale). Meno dell'1% dei punti DAPI non contengono YFP (corrispondente a non fluorescenti fagi) 5.
Time-lapse video:
Cellule litici sono riconosciuti da un accumulo di fluorescenza YFP (canale verde) all'interno della cellula, seguita da lisi cellulare. Lisogeni cellule sono riconosciuti da un accumulo di uniforme mCherry fluorescenza (rosso) all'interno della cellula e la ripresa della normale crescita cellulare e la divisione. Cellule non infettate (o cellule in cui l'infezione è fallito) non presenta alcuna delle fenotipi sopra e crescono e si dividono normalmente. Figura 7 mostra alcuni insiemi di immagini a contrasto di fase, YFP e canali mCherry, e le immagini corrispondenti sovrapposti di questi tre canali, da una tipica time-lapse movie (sezione 6). I fagi singoli (punti verdi) sono chiaramente visibili al periodo di tempo iniziale (Figura 7A). Tipicamente, un numerodi fagi sono visti sulla superficie cellulare (presumibilmente infettare le cellule), mentre altri sono fagi non adsorbito, come mostrato nella Figura 7B (pannello sinistro). Il risultato infezione diventa distinguibile nel tempo. Il ciclo litico è indicata dalla produzione intracellulare di nuovi fagi (verde, Figura 7C) seguita da lisi cellulare (cellule esploso con fagi Liberatoria verdi, figura 7D). Lisogenia è indicata dalla produzione di mCherry dal promotore RE P (rosso, Figura 7C) e la ripresa della crescita e divisione cellulare (rosso, figura 7D).




Figura 1. AB). Il fago viene purificato attraverso una serie di passi (CL pannelli).

Figura 2. Placche fagiche. Placche di fagi fluorescenti (a sinistra) sono inferiori a quelle di tipo selvatico (destra) dopo incubazione le piastre per 12 ore a 37 ° C.

Figura 3. Bande fagi dopo ultracentrifugazione. A) Due bande sono visibili dopo ultracentrifugazione in gradiente di fase CsCl. Quello superiore corrisponde a detriti cellulari e capsidi fagiche vuote, la banda inferiore contiene il fago desiderato. A sinistra: fago fluorescente, a destra. Wild type B ) Una band fago singolo è visibile dopo ultracentrifugazione in gradiente di equilibrio CsCl. La band fago fluorescente (a sinistra) ha un colore, a fronte di una banda blu per fago wild type (a destra).

Figura 4. La procedura di preparazione di lastre di gel di agarosio.

Figura 5. Immagini fluorescenti di fagi dopo colorazione DAPI. Fagi individuali sono facilmente distinguibili, e YFP e DAPI segnala co-localizzano molto bene.

Figura 6. Descrizione schematica di infezione fagica e la configurazione di imaging. Clicca qui per vedere una full-sized versione di questa immagine.
gura 7 "src =" / files/ftp_upload/3363/3363fig7.jpg "/>
Figura 7. Immagini tipiche di un time-lapse film di infezione fagica. Vengono mostrati il contrasto di fase, YFP e canali mCherry, così come una sovrapposizione dei tre canali. (A) YFP-canale immagini dal lasso di tempo iniziale. Sinistra, la somma di immagini YFP a z diversi posizioni. Le tre immagini di destra sono immagini campione YFP a z diversi posizioni, corrispondenti alle diverse zone della superficie cellulare. (B), (C) e (D) le immagini sovrapposti (a sinistra) del contrasto di fase (centro-sinistra), YFP (centro-destra) e mCherry (a destra) canali a tempi diversi. (B) a T = 0, si vedono due celle, ciascuna infettata da un singolo fago (punti verdi), ed una cellula viene infettata da 3 fagi. Anche osservato sono alcuni fagi adsorbito. (C) a T = 80 min, le due cellule infettate da fagi singoli sono andati in ciascun percorso litico, come indicanod dalla produzione intracellulare di nuovi fagi (verde). La cellula infettata da 3 fagi è andato in via lisogena, come indicato dalla produzione di mCherry dal promotore PRE (rosso). (D) A t = 2 h, il percorso litico ha portato a lisi cellulare (cellule esploso), mentre la cella lisogena ha diviso §.
§ Pannelli di sinistra di figura 7 (C) e (D) sono ristampati dalla cella, 141, Lanying Zeng, Samuel O. Skinner, Chenghang Zong, Jean Sippy, Michael Feiss, e Ido Golding, il processo decisionale a livello subcellulare determina il risultato di infezione batteriofago, 682-691, Copyright (2010), con il permesso di Elsevier.
| Scolare nome | Genotipo Rilevante | Fonte / riferimento |
| Ceppi batterici | ||
| LE392 | cenareF | John Cronan, University of Illinois |
| Fagi ceppi | ||
| λ LZ1 | GPD-EYFP, cI857 SAM7 D-EYFP b :: Kanr | Zeng et al. 5 |
| λ LZ2 | GPD-mosaico, genotipo come λ LZ1 | Zeng et al. 5 |
| Plasmidi | ||
| pP RE - mCherry | mCherry sotto il controllo di P RE, R amp | Zeng et al. 5 |
| pPLate * D | gpd sotto il controllo del promotore λ ritardo, amplificatore R | Zeng et al. 5 |
Tabella 1. Ceppi batterici,fagi e plasmidi utilizzati in questo lavoro.
| Ρ densità (g / ml) | CsCl (g) | SM (ml) | Indice di rifrazione η |
| 1,30 | 39 | 86 | 1,3625 |
| 1,50 | 67 | 82 | 1,3815 |
| 1,70 | 95 | 75 | 1,3990 |
Tabella 3. CsCl soluzioni preparate in tampone SM (100 ml) per gradienti passo.
Discussion
I ceppi batterici, fagi e plasmidi:
Strain LE392 è supF. E 'stato scelto per sopprimere la SAM7 mutazione nel genoma del fago (vedi Tabella 1 per i dettagli). Così, alla fine lysogens indotte lisi e rilasciare particelle fagiche, le cellule infettate volontà che hanno scelto la via litico. Lisogeni cellule vengono coltivate a 30 ° C a causa della presenza del termosensibile cI 857 allele nel genoma fagico. Dopo l'induzione di calore, GPD-EYFP e di tipo selvatico GPD sono co-espressi dal genoma di λ LZ1 e il plasmide pPlate * D rispettivamente. Come risultato, il capside del fago λ appena creato LZ2 contiene una miscela di GPD-EYFP e proteine GPD. Questo fago mosaico è strutturalmente stabile e sufficientemente fluorescente per consentire l'individuazione di fagi individuali 5. pP RE - mCherry è un plasmide reporter utilizzato per rilevare scelta del pathwa lisogenay. Il RE promotore P viene attivato dal CII durante la creazione di lisogenia 1,11. pP RE - mCherry 5 è stato derivato da pE-GFP 11 sostituendo GFP con mCherry 12. Per maggiori dettagli si veda il nostro precedente lavoro 5.
Condizioni di crescita Parametri:
Durante l'induzione lysogen (sezione 1), lieve agitazione a 180 rpm dà una resa virus buono 13. Uso di glucosio nel mezzo di crescita deve essere evitato il metabolismo del glucosio genera acidi prodotti metabolici, e particelle lambda mature sono instabili a pH acido 13. L'aggiunta di MgSO4 mira a stabilizzare il fago capside 3. Per i fagi che trasportano wild type CI (invece di CI 857), il lysogen può essere indotta con il DNA-danneggiamento agente Mitomicina C 3. Nella fase 1.3, l'incubazione a 37 ° C non dovrebbe di norma superare i 90 minuti. È Usef ul per verificare la densità cellulare da OD 600 ogni 30 min. Per un buon lisato, OD 600 scende a circa 0,2 o meno, e il restante OD 600 è un risultato di detriti cellulari. Incubazione troppo lungo può causare una resa inferiore, poiché il fago fago appena creato può iniziare a iniettare loro DNA in detriti cellulari. Per ottenere una banda visibile fago (almeno 1 x 10 11 particelle fagiche) nei passi 2,11 e 2,13, crescono almeno 500 ml cultura nel passaggio 1,2. L'aggiunta di 0,2% maltosio nel mezzo di crescita nelle fasi 5.1 e 5.2 mira ad indurre l'espressione di agnello, il recettore per il fago lambda adsorbimento 3,14. Il 1000 volte diluizione invece di 100 volte al punto 5.2 è volta a ridurre il livello di fondo mCherry dal RE plasmide giornalista pP - mCherry. Nel Passaggio 5,5 iniettabile DNA fagico triggering, 35 ° C viene scelta per evitare l'induzione della temperatura sensibile allele cI857.
Phage Purificazione:
jove_content "> Le fasi di depurazione dei fagi (passaggi da 2.1 a 2.11) può essere sostituito con altri 5 protocolli di purificazione, ma il finale attraverso ultracentrifugazione gradiente di equilibrio CsCl (Piazza di 2.12 e 2.13), è inevitabile. Rotori oscillanti sono necessari passi 2,10 e 2.12 per garantire taglienti bande fagiche visibili. Ottenere un titolo puro fago può facilmente richiedere fino a una settimana, per cui è necessario controllare il titolo fago lungo la strada per assicurarsi che nulla vada storto durante le fasi intermedie.Phage Manipolazione:
Durante tutte le procedure di purificazione di cui alla sezione 2, è fondamentale per gestire fago lisato con delicatezza per evitare di taglio code fago da teste fagiche. Durante l'infezione delle cellule nella sezione 5 (ad esempio, attraverso i passaggi 5,5 5,7), è anche fondamentale per evitare il taglio di particelle fagiche dalla cellula infettata. Si noti che se il fago viene tagliato dalla cellula infetta dopo iniezione suo DNA, il risultato è una infezione "scuro", cioè inrisultato perfezione sarà osservato nell'esperimento, ma il fago infettante non lo farà. Per ridurre al minimo tali problemi, si usa una punta larga pipetta ogni volta che la gestione fagi o il fago / cella miscela.
DAPI di prova:
La colorazione dello stock del fago con DAPI (sezione 4) è un metodo rapido ed efficace per verificare la purezza dello stock fago. Può anche essere usato per testare possibile degradazione di un magazzino fago esistente nel tempo. Per un archivio pura, la co-localizzazione di segnali YFP e DAPI al microscopio a fluorescenza dovrebbe essere prossimo al 100%. Abbiamo tipicamente osservare che meno dell'1% dei punti YFP non contengono DAPI (che rappresenta capsidi senza genoma virale), che indica che queste particelle non correttamente impacchettare il DNA virale o già iniettato loro DNA altrove. Meno dell'1% dei punti DAPI non contengono YFP (corrispondente a non fluorescenti fagi). Se questo non è il caso, attraverso passaggi 2,12 2,14 necessità di essere ripetuto in order per purificare nuovamente. Per quanto riguarda i parametri di imaging, la configurazione microscopio al punto 4.3 non è così critica come al punto 5 perché non a lungo termine live-cell imaging è necessario qui. Tuttavia, mantenendo le stesse impostazioni microscopia in sezione 5 è utile se si vuole calibrare l'intensità di fluorescenza di una singola particella fagica. Se il PBS-agarosio lastra non è molto pulito, o troppo colorante DAPI viene utilizzato, alcuni punti DAPI corrispondenti a DNA del fago può essere circondato da un "alone". Se troppo poco colorante DAPI viene utilizzato, il segnale dal canale DAPI può essere molto debole.
Microscopio di sistema:
Per l'imaging nella sezione 6, si usa un microscopio a epifluorescenza invertito commerciale (Eclipse TE2000-E, Nikon), con un obiettivo 100x (Piano di Fluo, apertura numerica 1,40, immersione in olio) e standard di set di filtri (Nikon). La sorgente di luce di fluorescenza è una lampada ad arco con controllo di intensità luminosa. Le seguenti caratteristiche sono controllati dal computer: x, y, z Pozione, campo chiaro e fluorescenza persiane, e la scelta del filtro di fluorescenza. Una funzione di messa a fuoco automatica è necessaria. In caso contrario, la messa a fuoco può facilmente allontanarsi durante il time-lapse film (normalmente 4 ore). La capacità di acquisire più posizioni (x, y) in ogni punto è utile, in quanto permette di seguire eventi di infezione multipli in parallelo. Noi di solito acquisire 8 posizioni stadio in ogni film, in seguito fino a 100 eventi di infezione. La fotocamera che usiamo è una soluzione raffreddata 512x512 CCD da 16x16 pixel fotocamera micron con una gamma dinamica di 16 bit (Cascade512, fotometrici). Acquisizione viene eseguita utilizzando MetaMorph software (Molecular Devices). Il microscopio deve essere posto in un ambiente a temperatura controllata, in alternativa, la fase microscopio dovrebbe essere circondato da una camera a temperatura controllata.
Acquisizione di immagini:
Per live-cell imaging, è fondamentale per evitare inutile esposizione del campione, che potrebbe portare a candeggio e phototoxicity. Pertanto, è meglio caratterizzare il vostro primo sistema per trovare una esposizione ottimale la luce che consenta di rilevare la fluorescenza pur non portando ad una crescita cellulare eccessiva sbiancamento o inibizione. Per ottenere una buona immagine di fluorescenza, giocare con l'intensità della luce eccitante, tempo di esposizione e guadagno della telecamera. Nei passaggi 6,2-6,3, l'intervallo di 10 min cornice è scelta allo scopo di minimizzare l'esposizione della luce. In ogni frame, un solo in-fuoco dell'immagine è necessaria in contrasto di fase (per riconoscimento cellulare) e canali fluorescenti (per determinare il destino della cellula). Nel primo punto temporale, tuttavia, più z-posizione immagini attraverso il canale YFP devono catturare tutti fagi infettano sulla superficie cellulare. Il tempo di esposizione YFP nel frame iniziale può anche essere superiore a quella utilizzata per il film accelerato nei tempi successivi.
Image Analysis:
Attentamente contare le particelle fagiche intorno alla superficie delle cellule nella fase 7.1. Comeosservato in precedenza, si prende una serie di z-stack attraverso il canale YFP al punto 6.2. Tuttavia, questo può ancora lasciare alcune particelle fagiche fluorescenti out-of-focus, che sfida il conteggio. La lunghezza della cella nel lasso di tempo iniziale è misurata utilizzando il software Metamorph. La lunghezza della cella può essere misurata ImageJ o altri strumenti software. Inoltre, un programma automatizzato casa costruita Matlab può essere molto utile per ottenere informazioni quali il cambiamento di fluorescenza nel tempo lungo linee cellulari.
Disclosures
Nessun conflitto di interessi dichiarati.
Acknowledgments
Siamo grati a Michael Feiss e Jean Sippy per la guida sulla creazione di fago e purificazione. Ringraziamo Michael Elowitz per la fornitura del software di riconoscimento delle cellule, Schnitzcell. Il lavoro in laboratorio Golding è sostenuta da sovvenzioni dal National Institutes of Health (R01GM082837), la National Science Foundation (082.265, PFC: Centro per la Fisica delle cellule viventi), la Fondazione Welch (Grant Q-1759) e Human Frontier Science Programma (RGY 70/2008).
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma-Aldrich | D4527-10KU | |
| RNase | Sigma-Aldrich | R4642-10MG | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| SM buffer | TEKnova, Inc. | S0249 | |
| NZYM | TEKnova, Inc. | N2062 | |
| CsCl | Sigma-Aldrich | C3011-250G | |
| Syringe | BD Biosciences | 309585 | |
| Needle | BD Biosciences | 305176 | |
| Dialysis cassette | Thermo Fisher Scientific, Inc. | 66333 | |
| Microscope slide | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti ultra-clear tube | Beckman Coulter Inc. | 344060 | |
| SW60Ti ultra-clear tube | Beckman Coulter Inc. | 344062 | |
| SW40Ti rotor | Beckman Coulter Inc. | 331302 | |
| SW60Ti rotor | Beckman Coulter Inc. | 335649 | |
| Refractometer | Fisher Scientific | 13-947 | |
| Epifluorescence microscope | Nikon Instruments | Eclipse TE2000-E | |
| Table 2. Reagents and equipment. | |||
References
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S.
- Ptashne, M. A genetic switch : phage lambda revisited. , 3rd edn, Cold Spring Harbor Laboratory Press. (2004).
- Hendrix, R. W. Lambda II. , Cold Spring Harbor Laboratory. (1983).
- Hershey, A. D. The Bacteriophage lambda. , Cold Spring Harbor Laboratory. (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. Current protocols in molecular biology. , John Wiley & Sons. (1994).
- Sambrook, J., Russell, D. W. Molecular cloning : a laboratory manual. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Fasman, G. D. Practical handbook of biochemistry and molecular biology. , CRC press. (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Personal communication with M. Feiss. , Forthcoming.
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).

{kind=link}