Motivation
Die Anstrengungen zur Verringerung der Nährstoffeinträge in Oberflächengewässer, die notwendig sind, unter anderemim Zusammenhang mit der Umsetzung der Europäischen Wasserrahmenrichtlinie1, erfordern eine genauere Prüfung der Phosphoremissionen. Ist der Stoffgruppe der Phosphonaten (Abbildung 1), die als Bleichmittel Stabilisatoren in Textil- und Papierindustrie, als Antiscalants in Trinkwasser-Aufbereitung, als Härte Stabilisatoren des Kühlwassers und in Wasch- und Reinigungsmitteln, verwendet werden besonders relevant in Bezug auf Quantität und Umweltrelevanz2. Phosphonaten werden verdächtigt, einen Beitrag zur langfristigen Eutrophierung von Wasser Körper2,3,4. Zum Beispiel durch UV-Strahlung des Sonnenlichts oder im Beisein von MnII und gelöster Sauerstoff, können Phosphonaten in mikrobiologisch verfügbaren Phosphate5,6abgebaut werden. Das Überangebot an Phosphat ist ein wesentliches Merkmal der ökologisch unausgewogen Gewässer, wodurch Phosphor eine wichtiges Ziel Substanz zur nachhaltigen Verbesserung des ökologischen Zustands der Gewässer.
Phosphonaten können durch Fällung/Flockung aus dem Abwasser entfernt, wenn mit Eisen oder Aluminium7,8,9,10 Salze. In diesem Prozess werden Metalle in kaum löslichen Metall-hydroxide verwandelt. Diese polaren Herden mit einer relativ großen spezifischen Oberfläche dienen als Adsorbentien für die negativ geladenen Phosphonaten. Die Flockung Prozess haben jedoch zwei Hauptnachteile. Je nach dem Abwasser können Schlamm Volumen von bis zu 30 % das Probenvolumen11auftreten. Dieser Schlamm muss getrennt, behandelt und in einem weiteren Sedimentation oder Filterstufe entsorgt werden. Darüber hinaus Phosphonaten können Komplex der zusätzlichen Flockungsmittel und somit verhindern die Bildung von Herden, vor allem im Abwasser mit geringer Wasserhärte. Dieser Effekt kann durch erhöhte Mengen an Flockungsmittel kompensiert werden. Dies führt jedoch zu erhöhten β-Werte (β = Molverhältnis von Flockungsmittel zu Phosphor im Abwasser)11,12. Eine komplexe Abwasser-Matrix kann daher die Kontrolle über eine optimale Flockungsmittel Dosierung erschweren.
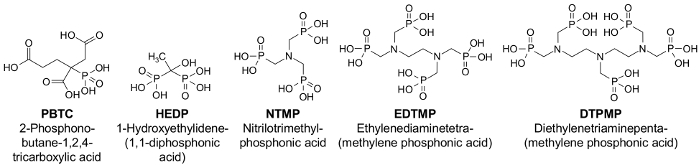
Abbildung 1: strukturelle Formeln wichtig Phosphonaten11. Bitte klicken Sie hier für eine größere Version dieser Figur.
Eine mögliche Alternative, die die hohe Adsorption Affinität von Phosphonaten metallhaltigen Oberflächen und nutzt hat keinen der oben genannten Nachteile sind Filtermaterialien basierend auf Eisenoxide (Hydr). Für solche Filtermaterialien präsentiert die Literatur vor allem Untersuchungen über die Eliminierung von Phosphat13,14,15,16. Dieses Papier stellt eine Prozedur ermöglicht die Untersuchung von der Aufnahmekapazität der selektiven granulierten Filtermaterialien in dieser Arbeit vor allem mit granular ferric Hydroxid (GFH), bezüglich Phosphonaten mit wenig Arbeitsaufwand und erhebliche Kosten zu sparen. Die Studie über die Aufnahmekapazität kann in folgende Schritte unterteilt werden: Vorbereitung der Phosphonat Lösung, Adsorption Test (Kontakt der Phosphonat-Lösung mit Granulat) und Phosphonat Analyse. Alle Schritte müssen perfekt aufeinander abgestimmt sein.
Konzept zur Adsorption Test und die Verwendung von geeigneten Puffern
Für das Studium der Aufnahmekapazität können Batch oder Spalte Tests durchgeführt werden. Ermittlung der Adsorption Isothermen oder pH-Abhängigkeiten von der Adsorbens ist der Batch-Ansatz bevorzugt, da viele Ergebnisse durch die Möglichkeit, mehrere Parameter variieren innerhalb kurzer Zeit erreicht werden können. Der pH-Wert ist einer der wichtigsten Einflussfaktoren Adsorption. Einhaltung bzw. Anpassung des pH-Wertes ist eine große Herausforderung für den Labortechniker, da die einfache Einstellung des pH-Wertes in der Probenlösung zuvor auf den Kontakt mit dem Adsorptionsmittel in der Regel nicht ausreicht. Jedes Adsorbens Material ist in der Regel bestrebt, den pH-Wert um diesen Punkt Null kostenlos (PZC) ungefähre. Dementsprechend ist es möglich, dass eine wässrige Lösung, z. B.auf pH 3 eingestellt auf einen pH-Wert von 8 im unmittelbaren Kontakt mit dem Adsorbens ändert. Abwasser hat meist eine natürliche Pufferkapazität, die diesen Effekt vermindert. Wenn nur die Entfernung eines bestimmten Ziel-Stoffes jedoch ist mit einer speziellen Adsorbens untersucht werden, synthetisches Abwasser muss verwendet werden, d. h., reines Wasser, die speziell mit dem Ziel Substanz oder, z.B., wettbewerbsfähige gespickt ist Anionen. Im Gegensatz zu pulverförmige Adsorbentien, wo der pH Wert kann leicht instand zu halten in den gewünschten Bereich durch Zugabe von Säuren und Basen in der offenen rührgefäßes, keine pH-Einstellung in dieser Form möglich ist, in einem Batch-Ansatz mit Granulaten. Um Granulat homogen ausgesetzt zu halten, sind sehr hohe Geschwindigkeiten rührende erforderlich, die in sehr schnellen Abrieb des Materials führen würde. Wenn solche Abrieb unbeabsichtigte ist, ist die sanfteste Methode, geschlossenen Zentrifuge Rohre, um das Granulat kontinuierlich in die Lösung gemischt zu halten rotieren. Der einzige Weg, um den pH-Wert konstant zu halten ist in diesem Fall, Puffer zu verwenden.
Die folgenden Anforderungen für Puffer erfüllt sein, um zu untersuchen, die Adsorption von Phosphat und Phosphonaten auf Eisen-haltige Filtermaterialien können: frei von Phosphor; farblos; löslich; im besten Fall keine Komplexbildner; keine Konkurrenz mit Phosphonaten hinsichtlich Adsorption auf polar Filtermaterialien; ähnliche Struktur der verschiedenen Puffer verwendet; und Puffer oder deren Abbauprodukte müssen keinen negativen Effekt auf die spektrale Absorption der Farbe Komplex nach der Verdauung für insgesamt P-Ermittlung. Bereich der biochemischen Forschung waren für so genannte gute Puffer entwickelten17,18,19, die genau diese Eigenschaften haben. So wurden für die Untersuchungen dieser Arbeit, die Puffer in der Tabelle 1 ausgewählt. Die pKeinen Wert von jeder Puffer zeigt den Bereich, der durch den Puffer konstant gehalten werden kann. Für die pH-Bereich < 5 müssen jedoch organische Säuren wie Zitronensäure (CitOH) und Essigsäure (AcOH) verwendet werden. Zitronensäure ist ein Komplexbildner, aber es puffert in einem pH-Bereich, wo die meisten eisenhaltigen Filtermaterialien sowieso instabil werden. Essigsäure und MOPS wurden bereits Nowack und Stein7 zur Adsorption von NTMP auf Gülle goethitgruppen (α-FeOOH) bei pH 4.6 und 7.2 zu untersuchen. Ihre Experimente auf der pH-Abhängigkeit der Adsorption erfolgte jedoch ohne Pufferung.
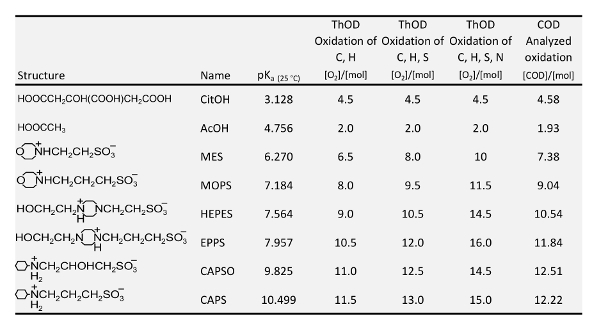
Tabelle 1: pK ein Werte 20 , Theoretischer Sauerstoffbedarf (ThSB) und analysiert aktuelle chemische Sauerstoffbedarf (CSB) des in dieser Studie verwendeten Puffer.
Gesamt-P-Ermittlung (ISOMini) angepasst an die Pufferlösung
Nach jedem Test Adsorption muss jede Lösung für die verbleibenden Phosphonat-Konzentration analysiert werden. Erst kürzlich wurde eine Methode zur Bestimmung von Phosphonaten in Umweltproben mit Grenzen der Quantifizierung im Bereich von 0,1 µg/L eingeführt. Es basiert auf der IC-ICP-MS-Methode und die Verwendung von Kationen-Austauscher (für die Umwandlung von Phosphonaten in “freien” phosphonic Säuren) und Anion Exchanger (für die Pre-Konzentration von Phosphonaten)21. Darüber hinaus wurde bereits im Jahr 1997 eine Methode von Nowack22 mit höheren Limits der Erkennung von 15-100 µg/L, eingeführt basiert auf der Pre-Komplexierung von Phosphonaten mit FeIII, Aufbewahrung mittels HPLC und den photometrischen Nachweis dieser -komplexe. Diese Methoden sind jedoch sehr aufwendig und teuer. In Studien mit synthetischen Abwasser, in denen die einzige phosphorhaltigen Substanz eine Phosphonat ist, ist es ausreichend, die Phosphonat-Konzentration zu bestimmen, durch die insgesamt P Konzentrationsbestimmung. Die Bestimmung von anorganischem Phosphat stellt den Experimentator mit weit weniger Probleme als die Bestimmung des Gesamt-P, wie letztere vorherigen Verdauung erfordert. Die Menge der Chemikalien, die pyramidenerbauer hinzukommen muss genau zu den Verbindungen in der Probe angepasst werden.
Die Bestimmung von Phosphat wird derzeit vor allem mit der Methode von Murphy und Riley23eingeführten durchgeführt. Diese Methode basiert auf die photometrische Erkennung von einem komplexen farbintensive Phosphomolybdenum blau ([PSb2Mo12O40]− mit λMax. bei 880 nm) die gebildet wird, in Anwesenheit von Phosphat und gesäuerte Molybdat mit Ascorbinsäure und antimony(III) als Reduktionsmittel24. In anderen Studien, das optimale Verhältnis von [H]+]: [Mo] war entschlossen, 60-8025,26. Um festzustellen, Gesamt-P, Verdauung, d. h., das Brechen des P-O-P, C-O-P und C-P Anleihen in phosphorhaltigen Verbindungen und die Oxidation von Phosphor zu Phosphat vorgenommen werden vor der Phosphomolybdenum blaue Bildung24 . Eisenreich Et al. 27 präsentiert eine vereinfachte Methode basiert auf der Verwendung von oxidierenden Agent disulfat (K2S2O8) im sauren Milieu. Viele dieser Erkenntnisse sind in die Entwicklung von ISO 687828, die systematisch, das Verfahren für die Bestimmung von Phosphat-P und total P-Konzentrationen in Wasserproben (Abwasser und Meerwasser erklärt) eingeflossen.
Die gesamte P Bestimmung gemäß ISO 6878 (Abbildung 2) erfordert die Probe in einen Erlenmeyerkolben von K2S2O8 bei einem sauren pH-Wert (Verwendung von Schwefelsäure) für mindestens 30 min verdaut werden. Nach der Verdauung soll der pH-Wert von 3-10 mit NaOH und den Inhalt der Erlenmeyer-Kolben in ein 50 mL volumetrischen Kolben übertragen wird. In dieser Flasche Ascorbinsäure und eine saure Lösung mit Molybdat und Antimon hinzugefügt, um die Probe und dann mit Wasser gefüllt. Nach 10-30 Minuten, die Intensität dieser blaue Färbung wird gemessen, bei einer Wellenlänge von 880 nm. Im Falle von Phosphat Bestimmung entfällt die Verdauung. Das bedeutet, die Probe wird in einem 50 mL volumetrischen Kolben mit Ascorbinsäure und eine Lösung mit Molybdat sowie Antimon gemischt, und die Intensität der Blaufärbung in das Photometer gemessen wird.
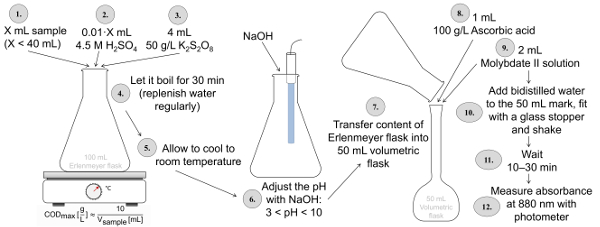
Abbildung 2 : Verfahren der insgesamt P Bestimmung gemäß ISO 6878 Anwendung Verdauung mit Schwefelsäure und Kalium disulfat, eine anschließende pH-Einstellung mit NaOH und Färbung mit Ascorbinsäure und Molybdat-haltigen Lösungen. Bitte klicken Sie hier für eine größere Version dieser Figur.
Das Verfahren des gesamten P-Ermittlung ist sehr komplex, da während der Verdauung, die es immer, die achten muss die Probe nicht über Kochen und die Anpassung der Probe auf pH 3-10 sehr lange dauert. Um möglichst viele Proben wie möglich in kürzester Zeit analysieren zu können, wurde eine miniaturisierte Form des Gesamt-P und ortho-Phosphat-Bestimmung anhand dieser ISO-Methode entwickelt. Abbildung 3 fasst die einzelnen Schritte dieser Methode. In dieser miniaturisierten Entschlossenheit Methode (ISOMini), ist der letzte Band der Farbe Lösung 10 mL (Dies ist in der ISO-Methode, 50 mL). Entsprechend reduziert die ISO-Mini -Methode die Menge der Lösungen auf ein Fünftel verwendet werden. In der ISO-Mini -Methode die Verdauung erfolgt in einem Thermostat (im Gegensatz zu den ISO-Methode, wo die Verdauung in einen Erlenmeyerkolben auf einer Herdplatte vorgeschlagen wird) 148-150 ° c, die höchste mögliche Oxidation zu erhalten. NaOH wird nach der Verdauung zusammen mit Ascorbinsäure und sauren Molybdat Lösung hinzugefügt.
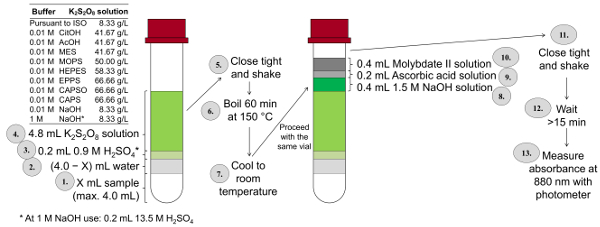
Abbildung 3 : Verfahren der gesamten P Bestimmung nach einem modifizierten und miniaturisierte Form des ISO 6878 (ISOMini) mit Schraubkappe 10 mL Fläschchen, Puffer-abhängigen Kalium disulfat Konzentrationen, Heizung Thermostat und Zugabe von Farbe Reagenzien direkt an die verdaute Probe ohne Sie vorher zu übertragen. Bitte klicken Sie hier für eine größere Version dieser Figur.
Die biologische Puffer enthalten in den Proben müssen in relativ hohen Konzentrationen (10 mM) im Vergleich zu den Phosphonat (5-30 µM) vorhanden sein, um den pH Wert effektiv erhalten. Diese Puffer müssen nach der Adsorption-Test zur Analyse des gesamten P verdaut werden. Dementsprechend muss die dosierte Menge des Oxidationsmittels in jeder Puffer, unter Berücksichtigung, dass zuviel Oxidationsmittel die Bildung von komplexen Farbe gebildet nach der Verdauung nicht beeinträchtigen sollte angepasst werden. Um die K2S2O8 benötigte Menge für die Verdauung von jeder Puffer bei der gesamten P-Bestimmung anhand der analysierten chemischen Sauerstoffbedarf (CSB) abschätzen zu können, ein Vergleich, wie viele Elektronen umgewandelt werden kann, während die Reduzierung der O2 und K2S2O8 ist erforderlich:
O2 + 4 H+ + 4 e– → 2 H2O
S2O82- + 2 e– → 2 SO42-
Daher erfordert die Oxidation eines bestimmten Moleküls doppelt soviele disulfat Moleküle als O2 Moleküle. Dementsprechend muss bei ein Probenvolumen von 20 mL COD der Probe nicht 500 mg/L überschreiten bei Verwendung der ISO-Methode. Allerdings ist auch bei MES, der gute Puffer mit der kleinsten molare Masse von Tabelle 1, bereits ein DORSCH von 2,4 g/L bei einer Konzentration von 10 mM. Neben der schrittweise Protokoll der Adsorption Test- und ISOMini Methode, dieses Papier untersucht daher die erforderlichen Puffer-Konzentration, der Einfluss der Puffer auf Phosphonat Adsorption und K2S2O8 Menge und NaOH Dosierung für ihre Verdauung in der ISO-Mini -Methode erforderlich.
Freundlich-Modell der adsorption
Adsorption Isothermen, d. h.be-Q (z. B.in mg P/g Adsorbens) über die gelöste Konzentration c (in mg/L P) der adsorptiven nach einer bestimmten Einwirkzeit angewendet kann mit der Gleichung von Freundlich29vorgeschlagenen modelliert werden:

Wenn die experimentell ermittelten Werte Q und c in Form von einer Funktion ln(q) über ln(c) aufgetragen sind, entspricht die Steigung dieser Funktion durch lineare Regression bestimmt 1/n und die y-Achse abfangen, KF Wert30.
Überblick über das Verfahren
Der gesamte Prozess zur Ermittlung der Aufnahmekapazität der granular ferric Hydroxid in Bezug auf Phosphonaten ist in mehrere Schritte unterteilt und wird im Abschnitt Protokoll beschrieben. Für die Analyse ist es notwendig, eine ausreichende Menge an Reagenzlösungen (Abschnitt 1 des Protokolls) vorzubereiten. Diese sind für mehrere Wochen haltbar. Die Phosphonat-haltige Lösung ist dann bereit (Abschnitt 2), gefolgt von der Adsorption-Test (Kontakt der Phosphonat-Lösung mit dem Granulat) (Abschnitt 3) und die Analyse des gesamten P nach der miniaturisierten ISO-Methode (Abschnitt 4).
