Multiple fluorescent reporters can be used to measure exophers. Touch neuron exophers are readily visualized in vivo via fluorescent tagging of proteins that may be selected for extrusion, by labelling of organelles that can be extruded, or by tagging cell membranes. Table 1 identifies touch neuron expressed fluorescent reporters that have been used to monitor exophers, with representative examples included in Figure 4. Cargoes that are known to be extruded in exophers include a fusion of the N-terminal domain of human huntingtin to expanded polyglutamine (Q128) (Figure 4B), lysosomes that are GFP-tagged with lysosomal associated membrane protein (LMP-1) (Figure 4C), and mitochondria tagged with matrix-localized GFP (Figure 4D). Cytoplasmic GFP is not strongly expelled and is preferentially retained in the soma5, although GFP can weakly visualize exophers (Figure 4A). When GFP is fused to proteins that are expelled, this tag can be used to visualize exophers. An important point is that by tagging different proteins, a large range of questions on the expulsion of specific cargoes and organelles, as well as on the proteins and membranes that make up exophers, can be addressed.
A pseudo-stereomicroscope setup is an effective tool for viewing exophers in animals upon agar plates. This setup is a hybrid of compound and stereoscopic technology that includes high numerical aperture optics on each magnification, pseudo-stereo technology (discrete objectives over a stereoscopic base), and a zoom operating switch for viewing at magnifications intermediate to installed objectives. A microscope such as this should be equipped with 10x eyepieces and objectives powerful enough for observing neuronal morphology and exopher production for high-throughput scoring (2x objective used for scanning/picking, 10x objective used for identifying and scoring).
While magnification capabilities of standard stereomicroscopes typically have high enough resolution to see the network of touch neurons expressing fluorescent proteins, standard dissecting microscopes are not sufficient for observing subcellular details of exophers like the tubular connections of soma to exopher. Such observations necessitate confocal microscopy (see the Table of Materials for equipment details).
Exopher quantitation studies require strict controls to eliminate experimental stresses. The attentive maintenance of consistent growth conditions is required for reproducible exopher production. More specifically, exopher production is stress-responsive such that consistent feeding, constant temperature, and contamination-free growth across generations are critical for reproducibility. Under basal growth conditions with high neuronal expression of mCherry, exopher production is relatively low (5-25% of ALMRs produce exophers) but some stresses, including osmotic and oxidative stress, can increase exopher rates. While mCherry expression can be thought of as stress, a corollary of the stress-sensitivity of exopher levels is that, if properly controlled, experimental stress introduction can be a strategy to more easily induce and observe exophergenesis.
Timing and anticipated exopher production levels. Exophers are virtually absent during larval development. The period of peak exopher production in young adult life appears to be highly restricted to during adult days 1-4, most commonly being evident at adult day 2 or 3. Because the peak can shift ahead or back a little, the most complete evaluation of an exopher production profile is to score multiple trials daily over adult days 1-4. In general, an ALMR produces one major exopher, with the vesicle persisting for at least 24 hours. The exopher can be produced fairly quickly (on the order of minutes at its fastest). Most commonly, only one major exopher is produced per neuron in early adult life, but production of multiple exophers is possible.
In general exopher production by ALMRs expressing mCherry under basal conditions ranges from 5-25% of ALMRs examined within the optimal timeframe of adult day 2-3 (Figure 3D). Proteostasis crises5, as well as exposure to other stresses can modulate exopher level. Stress or genetic perturbations can increase exopher production to detection rates as high as 90% of ALMR neurons producing exopher extrusions.
Feeding-based RNAi for testing roles of specific genes in exophergenesis. The nematode C. elegans is commonly subjected to RNAi knock down by feeding animals transformed E. coli strain HT115 that express a double stranded RNA (dsRNA) targeting a gene of interest20. HT115 bacteria can be used when scoring for exophers in feeding RNAi5. While transcripts in most tissues can be targeted by RNAi using this technique, neurons are more refractory. Sensitivity to RNAi can be calibrated using animals that express the transgenic dsRNA transporter SID-1 under a neuron-specific promoter. In this way neuronal tissue can be sensitized to RNAi21.
Tissue-specific knockdown of a gene of interest can be accomplished by expressing a component of endogenous RNAi metabolism within a mutant that is deficient in that component. For example: the Argonaute protein RDE-1 can be expressed specifically in the neurons of rde-1 mutant animals to achieve knockdown of a gene of interest only in neurons when animals are exposed to an RNAi intervention targeting that gene.
Using standard nematode RNAi protocols20,22, exposure of the parents at the L4 stage to the RNAi and allowing their progeny to develop consuming transformed HT115 bacteria until adulthood generates the strong genetic knock-down but be attentive to potential developmental delays induced by RNAi as experimental animals may grow differently than an empty vector control. It is important to always include the empty vector control for negative control comparison. HT115 bacteria can be used when scoring for exophers in feeding RNAi. However, note that some genes are effective at changing exophergenesis rates even during shorter periods of RNAi exposure5. If targeting of certain genes leads to developmental failure, avoid exposing animals to lifelong knockdown, animals can simply be picked at the L4 stage onto RNAi plates for exposure from L4 to adult D2 or D3.
| Strain name | Genotype | Description | Exopher percentage | Reference |
| SK4005 | zdIs5[Pmec-4GFP] | Cytosolic expression of GFP in touch neurons. | 1-8% ALM | Figure 4A, Melentijevic 2017 |
| ZB4065 | bzIs166[Pmec-4::mCherry] | Overexpression of mCherry (bzIs166) in touch neurons, produces both cytosolic signal and mCherry aggregates. bzIs166 is an exopher inducer. mCherry aggregates are predictors of exophergenesis and are preferentially extruded in exophers. | 3-20% ALM (normal conditions). 20-80% ALM (fasting conditions). | Figure 4B, Melentijevic 2017 |
| ZB4067 | bzIs167[Pmec-4mitogfp Pmec-4mCherry4]; igIs1[Pmec-7YFP Pmec-3htt57Q128::cfp lin-15+]; | YFP cytosolically labels mec-7 touch neurons. Co-expressed Q128::CFP aggregates and induces exophers. CFP preferentially silences. | ~25% | Figure 4C, Meletijevic 2017 |
| ZB4509 | bzIs166[Pmec-4mCherry]; bzIs168[Pmec-7LMP-1::GFP] | bzIs168 LMP-1::GFP labels plasma membranes and lysosomal membranes. bzIs168 can be used to identify neuronal membranes, exophers (as they are membrane bound), and lysosomal-membrane structures. | 3-20% ALM | Figure 4D, Melentijevic 2017 |
| ZB4528 | bzIs166[Pmec-4mCherry]; zhsEx17 [Pmec-4mitoLS::ROGFP] | Allele zhsEx17 is a mitochondrially localized reporter that changes its peak excitation wavelength from 405nm (oxidized) to 476nm (reduced) according to the local oxidative environment. It is expressed in the touch neurons and can be used on its own to identify mitochondria in touch neurons and in mito-exophers. | 3-20% ALM proteo-exopher. % ALM mito-exopher quantitation in progress. | Figure 4E, Melentijevic 2017, Cannon 2008, Ghose 2013 |
Table 1. Strains that have been used for visualization of touch neurons, touch neuron-exophers, and exopher contents.
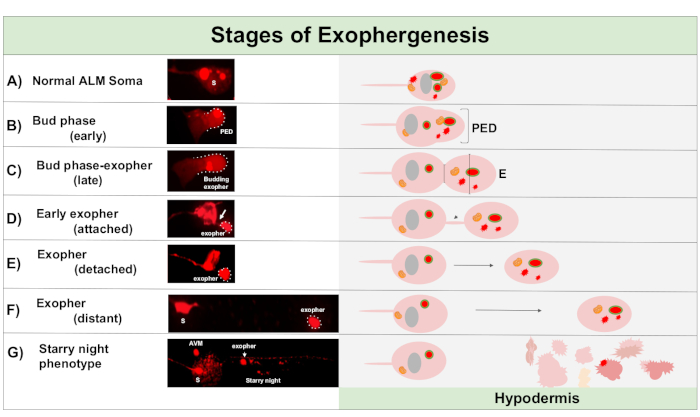
Figure 1: Stages of Exophergenesis. The process of making and ejecting an exopher is called ‘exopher-genesis’. The dynamic process of exopher formation can take several minutes to several hours. Depicted are examples of soma and exopher morphology at specific steps during the dynamic exophergenesis process in a high-exopher producing strain, ZB4065 bzIs166[Pmec-4mCherry]. All images are of day 2 adult ALM neurons taken with a 100x objective. (A) Normal soma. Adult mechanosensory touch neuron ALM transgenically expressing Pmec-4mCherry. The soma morphology depicted is typical of young adult neurons in this strain, with mCherry concentrations in the cytoplasm. (B) Early bud phase. The first observable step of exophergenesis involves polarization of selected cytoplasmic material to the edge of the soma membrane. This step is often accompanied by an expansion or swelling of the soma. In the case of the touch neurons, the pre-exopher domain (PED) extends into the surrounding hypodermis (not visible here). Note the greater concentration of mCherry material into the early bud domain. (C) Late bud phase. Upon further cellular polarization and an expansion of the pre-exopher domain, a constriction between the soma and exopher (arrow) becomes evident. This event signals the transition to the late bud phase. Although in the late bud stage the cell exhibits a clear constriction site and separate soma and exopher domains, it is not yet pinched off completely from the soma; the budding exopher may be attached by a thick stalk (arrow). The budding domain is considered an early exopher when the diameter of the exopher domain in question is roughly ⅓ larger than the diameter of the construction site/stalk. (D) Early-exopher phase. Early exophers can be attached by a stalk from the departing soma—the diameter of this connection can thin as the exopher moves away from the soma. Cytoplasmic material can be transferred from the soma to the exopher via this tube, although most material is loaded during the process of budding out. Exophers can detach from the soma as depicted in (E), separated exophers are considered mature exophers (F). The mature exopher can transit through the surrounding hypodermal tissue, moving away from the departing soma. (G) Breakdown of the mCherry-labelled exopher into smaller vesicles within the hypodermis results in a scattered punctate appearance of the mCherry material, most likely as it enters the hypodermal endolysosomal network. The dispersed punctate signal is called the “starry night” phase. Degradation of some exopher contents is likely accomplished by hypodermal lysosomes, but some material is not fully degraded and is often re-extruded by the hypodermis into the pseudocoelom. The post-exophergenesis mCherry transit is described in more detail in Figure 2. Please click here to view a larger version of this figure.
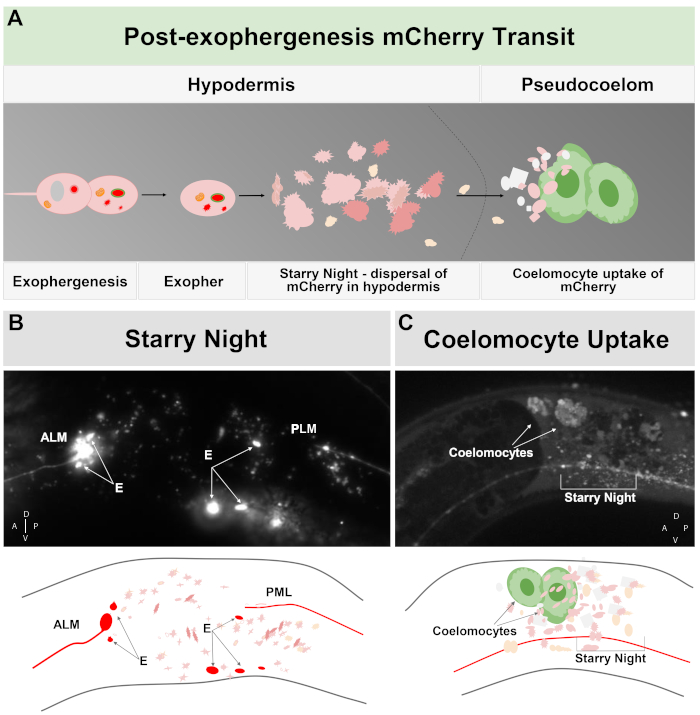
Figure 2: mCherry extruded from touch neurons in exophers engages the surrounding hypodermal lysosomal network but can later be extruded into the pseudocoelom where coelomocytes can store/degrade the mCherry. (A) Cartoon summary of how mCherry extruded in exophers transits the body after expulsion by neurons. During exophergenesis selected cellular contents such as mCherry become localized and bud off from the sending neuronal soma in an independent vesicle surrounded by the neuronal and hypodermal plasma membranes. Since the touch neurons are embedded in the hypodermal tissue, as the exopher domain buds outwards it moves further into the hypodermis. The exopher can transit the hypodermis, and after hours to days, exopher contents can fragment within the endolysosomal network of the hypodermis. The mCherry can appear as scattered puncta throughout the hypodermis, a stage called “starry night”. After a few days, some of the mCherry can pass out of the hypodermis into the surrounding pseudocoelom, where scavenger cells called coelomocytes can get access to, and take up, mCherry that can be stored. (B) Example of the appearance of the starry night mCherry vesicles. Image of an ALM soma tagged with mCherry with large exopher fragments and starry night vesicles. Strain is ZB4065 bzIs166[Pmec-4mCherry]. (C) Example of mCherry concentration in distant coelomocytes. Sideview of an adult animal day 10 of strain ZB4065 bzIs166[Pmec-4mCherry] showing mCherry concentrated in coelomocytes (arrows). Some starry night vesicles are also evident. In general coelomocyte concentration becomes evident after about adult day 5 of life. (B bottom) Cartoon reproduction of (B), with touch neurons and processes outlined in red, as are brightest exopher fragments; scattered small vesicles of different Z-depths are shown in lighter pink. (C bottom) Cartoon version of image of (C), showing neuronal process in red, starry night in pink and coelomocytes in green. Please click here to view a larger version of this figure.

Figure 3: Mechanosensory touch neurons produce exophers at different levels with a precise temporal profile. (A) (Top) Cartoon depiction of mechanosensory touch neurons in spatial relation to key anatomical landmarks of C. elegans including the pumping pharynx and the neuron-dense nerve ring at the head of the animal, the vulva at the mid body, and the tapered tail. (Bottom) Fluorescently labeled touch neurons expressing GFP as viewed from the top and left side (images adapted from WormAtlas). The red box depicts the area where ALM exophers are typically located. (B) High magnification view of the mid body region at which ALM-derived exophers are produced in a strain expressing [Pmec-4mCherry]. AVM and ALMR neuron are depicted, and shown is an ALMR exopher along with mCherry starry night. ALMR neurons most readily produce exophers. (C) ALMR mechanosensory touch neurons more readily produce exophers compared to other touch neurons in hermaphrodites under basal conditions. Mechanosensory touch neuron exopher production on adult day 2, as scored for individual touch receptor neurons is indicated. Strain: ZB4065 bzIs166[Pmec-4mCherry], N>150, error bars are SEM. (D) ALMR touch neurons produce more exophers during days 2 and 3 of adulthood compared with the adolescent L4 stage or with animals in advanced age. Strain: ZB4065 bzIs166[Pmec-4mCherry], N>150, error bars are SEM. Please click here to view a larger version of this figure.

Figure 4: Examples of some fluorescent reporters that tag exopher contents. A straightforward way to observe exophers is by creating transgenic animals that express fluorophores from neuronal promoters. The fluorophores allow for visualization of the exopher and transgenic expression induces aggregation and/or proteostress that increases exophergenesis. Exophers produced by amphid neurons can also be observed under native conditions, using dye filling for visualization. Shown are examples of common strains that can be used to observe exophers, (E) exopher, (S) soma. (A) Soma and exopher from an ALM of an adult of strain SK4005 zdIs5[Pmec-4GFP],100x objective used for photography, scale bar 3μm. In this strain, exophers that include soluble GFP are measured, but exopher production occurs infrequently. Fusing GFP to proteins that can be preferentially extruded in exophers in other studies confirms that GFP fusions can be detected in mature exophers. (B) ALM soma and exopher of an adult of strain ZB4065 bzIs166[Pmec-4mCherry], which expresses mCherry and induces touch neuron exopher production. 100x objective used for photography, scale bar 5 μm. (C) ALM soma and exopher of an adult of strain ZB4067 bzIs167[Pmec-4mitogfp Pmec-4mCherry4]; igIs1[Pmec-7YFP Pmec-3htt57Q128::cfp lin-15+]; selective blue channel used for image of htt57Q128::CFP. The exopher contains htt57Q128::CFP aggregates (arrows), that appear more concentrated in the exopher than in the soma. 40x objective used for photography, scale bar 5μm. (D-E) Exophers can contain organelles and organelle-specific tagging with fluorescent proteins enables monitoring of organelle extrusion. (D) Lysosomal membrane tag LMP-1::GFP outlines the soma and exopher membrane and tags plasma membranes weakly (plasma membrane localization is a trafficking step on the way to lysosomal targeting) and labels lysosomal organelles strongly. Shown is an adult ALM soma co-expressing Pmec-4mCherry and the Pmec-7LMP-1::GFP that localizes to membranes and lysosomes. The soma has an attached exopher with other smaller extrusions likely to be exopher fragments (arrows). GFP positive structures are included in the soma and are present in the large exopher, strain: ZB4509 bzIs166[Pmec-4mCherry]; bzIs168[Pmec-7LMP-1::GFP]. 100x objective used for photography, scale bar 5 μm. E) A mitochondrial GFP marker can be used to identify mitochondria in soma and exophers. Shown is an adult ALM soma expressing Pmec-4mCherry and mito::ROGFP, which localizes to the mitochondrial matrix. mito::ROGFP expressed alone, without the mCherry, can also readily be used to identify neurons and score for exophers that include mitochondria. Strain: ZB4528 bzIs166[Pmec-4mCherry]; zhsEx17 [Pmec-4mitoLS::ROGFP]. 100x objective used for photography; scale bar 5μm. Please click here to view a larger version of this figure.

Figure 5: Developmental cycle of C. elegans and L4 identification. (A) At 20 °C an egg takes approximately 9 hours to hatch once laid by the mother. (B) A newly hatched animal is in larval stage 1 (L1) and molts into an L2 larva after 12 hours. (C) Animals remain in the L2 and the (D) L3 larval stages for about 8 hours each. (E) Adolescent animals are considered the fourth larval stage (L4) and are marked by a conspicuous developing vulva that appears as a white crescent near the mid body. The presence of this while crescent enables easy identification and picking of L4 staged animals to establish synchronized cultures that later facilitate scoring for exophers. Animals remain in the L4 stage for about 10 hours before their final molt into gravid adults, F) identified by developing eggs, visible spermatheca, and the initiation of egg-laying. Please click here to view a larger version of this figure.
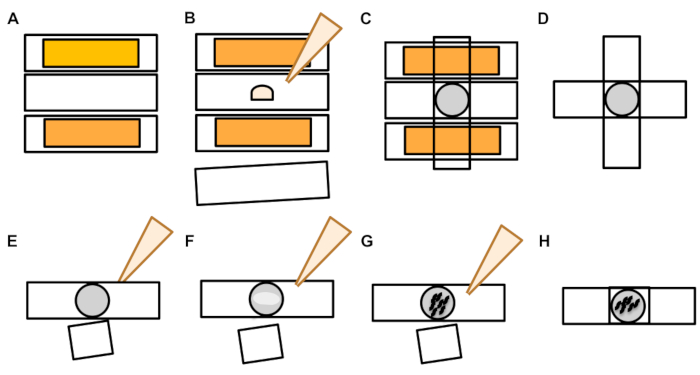
Figure 6: Preparation of microscope slide agar pad. (A) Prepare two slides with a single strip of laboratory tape placed lengthwise across the top. Place a non-taped microscope slide in between as pictured. B) Place a drop of molten agarose on top of the slide. (C) Place a clean slide gently on top of the drop, pressing the agarose into a deflated circle pad. (D) Remove the taped slides, which act to accomplish an even flattening of the agar that is needed to create an even pad. (E) Remove the top slide once the agarose pad has dried. (F) Pipette a paralytic solution (levamisole or tetramisole) on top of the agar pad. (G) Pick appropriately staged animals into the paralytic. (H) Gently cover the animals with a coverslip and ensure animals are alive. Please click here to view a larger version of this figure.

Figure 7: Characters of exophers and exopher identification criteria. (A) General criteria that identify an exopher. (B) Diameter comparisons between the sending soma and the extruded exopher, measured in μm. Adult ALM somas, N=35, strain: ZB4065 bzIs166[Pmec-4mCherry] – 6.53 μm average size of soma and 3.83 μm average size of exopher. (C) Defining criteria for differentiating between an exopher domain and a budding exopher. (D) Most commonly, individual neurons make one large exopher, which later splits or fragments as the hypodermis attempts to degrade its contents. Still, multiple exophers may be observed next to one touch neuron that might derive from either multiple exopher events from one neuron or alternatively, exophers can also bud or fragment themselves. The origin of multiple exopher-like entities can only be determined using time lapse microscopy. Top depicts an ALMR touch neuron soma with a single distant exopher. Bottom depicts an ALMR touch neuron soma with multiple exopher-like extrusions. (E) Common morphological features in adult ALM touch neuron somas that may be mistaken for exopher events. Top left – A distended ALM soma, with no clear exopher domain or constriction site. Top middle – Neurons can have small extracellular protrusions that may be analogous to exophers, but do not meet size requirement criteria to be considered an exopher. Top right – With age, touch neurons can develop outgrowths along their minor neurite. Often mCherry material can be collected at the tip of the neurite outgrowth. This is not scored as an exopher if the collected mCherry does not meet exopher-to-soma size requirements. Bottom depicts adult ALM neurons that have defining criteria for an exopher domain or an exopher. Botom left – ALM soma that has a prominent exopher domain that selectively includes mCherry cytosol and mCherry tagged aggregates. The exopher domain constriction site is marked by arrows and meets the size criteria (at least 1/5th the size of the soma). The largest diameter of the exopher domain is almost ⅓ bigger than the diameter of the constriction site, meeting criteria for an exopher event. Bottom middle – ALM soma that has a prominent budding exopher that meets the size criteria. There is a clear constriction site. Bottom right – ALM soma that has an attached mCherry-filled exopher that meets exopher size requirements. The exopher is attached by a thin connecting filament. All images are from strain ZB4065 bzIs166[Pmec-4mCherry]. Please click here to view a larger version of this figure.
