

Summary
A formação de feixes de actomiosina in vitro e a medição da geração de força do conjunto de miosina usando pinças ópticas são apresentadas e discutidas.
Abstract
As miosinas são proteínas motoras que hidrolisam o ATP para percorrer as trilhas do filamento de actina (FA) e são essenciais em processos celulares, como motilidade e contração muscular. Para entender seus mecanismos geradores de força, a miosina II tem sido investigada tanto no nível de molécula única (SM) quanto como equipes de motores in vitro usando métodos biofísicos, como armadilhas ópticas.
Esses estudos mostraram que o comportamento gerador de força da miosina pode diferir muito quando se move do nível de molécula única em um arranjo de três esferas para grupos de motores trabalhando juntos em um grânulo rígido ou superfície de deslizamento de cobertura em um arranjo de deslizamento. No entanto, essas construções de ensaio não permitem avaliar a dinâmica de grupo da miosina dentro da hierarquia estrutural viscoelástica como fariam dentro de uma célula. Desenvolvemos um método utilizando pinças ópticas para investigar a mecânica de geração de força por conjuntos de miosina interagindo com múltiplos filamentos de actina.
Esses feixes de actomiosina facilitam a investigação em um ambiente hierárquico e complacente que captura a comunicação motora e a saída de força do conjunto. A natureza personalizável do ensaio permite alterar as condições experimentais para entender como as modificações no conjunto de miosina, no feixe de filamentos de actina ou no ambiente circundante resultam em diferentes saídas de força.
Introduction
As proteínas motoras são essenciais à vida, convertendo energia química em trabalho mecânico 1,2,3. Os motores de miosina interagem com os filamentos de actina dando passos ao longo dos filamentos semelhantes a uma trilha, e a dinâmica das redes de actina-miosina realiza a contração muscular, a motilidade celular, o anel contrátil durante a citocinese e o movimento de carga dentro da célula, entre outras tarefas essenciais 3,4,5,6,7,8 . Como as miosinas têm tantos papéis essenciais, a falha na funcionalidade da rede miosina-actina pode levar ao desenvolvimento de doenças, como mutações na cadeia pesada da miosina que causam hipercontratilidade cardíaca na cardiomiopatia hipertrófica (CMH)9,10,11,12,13,14 . Na contração muscular, os motores individuais de miosina cooperam entre si, trabalhando como um conjunto para fornecer a energia mecânica necessária que realiza o deslizamento relativo das FAs 4,15,16,17,18. Os motores de miosina formam pontes transversais entre as FAs e utilizam alterações conformacionais devido ao seu ciclo mecanoquímico para se moverem coletivamente em direção à extremidade farpada dos filamentos alinhados 17,18,19,20,21.
O desenvolvimento de ensaios quantitativos de motilidade in vitro no nível do SM usando técnicas como o aprisionamento óptico facilitou a coleta de detalhes sem precedentes de como os motores individuais de miosina funcionam, incluindo a medição da geração de força SM e os tamanhos dos passos 22,23,24,25,26,27,28,29,30 . Finer et al. desenvolveram o ensaio de aprisionamento óptico "três contas" ou "halteres" para investigar a mecânica de geração de força de motores de miosina II simples23,31. Como a miosina II muscular trabalha em equipes para contrair FAs, mas não é processiva no nível do SM, a orientação do ensaio de aprisionamento óptico teve que ser rearranjada a partir da abordagem clássica do grânulo ligado ao motor32. Para formar o ensaio de halteres, duas armadilhas ópticas foram utilizadas para segurar uma FA sobre um motor de miosina ligado a um cordão preso por deslizamento de cobertura, e a saída de força pelo motor único foi medida através de movimentos da FA dentro da armadilha23.
No entanto, as forças SM e o uso de uma única orientação motor/ensaio de filamento único não fornecem uma imagem completa sobre a geração de força no nível do sistema, uma vez que muitas proteínas motoras, incluindo a miosina II, não funcionam isoladamente e muitas vezes não funcionam como uma soma de suas partes 15,16,17,32,33,34,35,36 . Estruturas mais complexas que incluam mais de um motor interagindo com mais de um filamento são necessárias para melhor compreender a sinergia das redes de filamentos de miosina e actina15,32. A orientação do ensaio de halteres tem sido explorada para investigar a geração de força de pequenos conjuntos por ter múltiplas miosinas ligadas a um grânulo ou usando um filamento de miosina espessa ligado a uma superfície e permitindo que os motores interajam com a FA suspensa 4,23,34,37,38,39,40.
Outros pequenos ensaios de conjunto incluem um ensaio de deslizamento de filamento in vitro em que os motores de miosina são revestidos em uma superfície de deslizamento de cobertura, e um talão ligado a um FA é usado para sondar a força gerada pela equipe de motores 4,35,36,38,39,40,41,42,43 . Em ambos os casos, as miosinas estão ligadas a uma superfície rígida – grânulos ou deslizamento de cobertura – e utilizam um AF. Nesses casos, os motores não são capazes de se mover livremente ou se comunicar uns com os outros, nem ter miosinas rigidamente ligadas reflete o ambiente hierárquico e compatível em que os motores trabalhariam juntos no sarcômero32. Estudos anteriores sugeriram que a miosina II pode sentir seu ambiente e adaptar-se de acordo com as mudanças nas condições de concentração viscoelástica ou motora, alterando características como geração de força e razão de serviço41,44,45. Assim, há uma necessidade de desenvolver um ensaio de armadilhagem óptica que promova e capture a comunicação motora e a conformidade com o sistema para pintar uma imagem mais realista dos fundamentos mecanicistas da geração de força do conjunto de miosina II.
Aqui, desenvolvemos um método para acoplar estrutura hierárquica in vitro com aprisionamento óptico, formando feixes de actomiosina ou sanduíches consistindo de múltiplos motores de miosina interagindo entre dois filamentos de actina. Esta geometria de ensaio modular tem a capacidade de investigar diretamente como os fatores moleculares e ambientais influenciam a geração de força de miosina do conjunto. Além disso, investigar os mecanismos de geração de força por meio desses conjuntos de actina-miosina tem o potencial de auxiliar na modelagem e compreensão de como tarefas celulares em larga escala, como a contração muscular, se propagam a partir do nível molecular 9,10,13.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Folhas de cobertura de gravação
- Dissolver 100 g de KOH em 300 mL de etanol a 100% em um copo de 1.000 mL. Mexa com uma barra de agitação até que a maioria do KOH tenha se dissolvido.
CUIDADO: A solução concentrada de KOH pode causar queimaduras e danos à roupa. Use luvas, proteção para os olhos e um jaleco. - Coloque as tampas individualmente em racks de limpeza de tampas.
NOTA: Os racks são projetados com fendas que mantêm as tampas simples espaçadas para permitir a gravação e o enxágue em cada face da tampa, os orifícios de drenagem no fundo e feitos de material que pode suportar as condições adversas de gravação. Eles podem ser feitos sob medida ou comprados comercialmente. - Preparar e rotular três copos de 1.000 mL: um com 300 mL de etanol e dois copos com 300 mL de água de osmose reversa (RO).
NOTA: Aqui, a água RO foi obtida de um purificador de água de laboratório, mas também pode ser comprada comercialmente se um purificador local não estiver disponível. - Coloque cada um dos quatro copos em um sonicator de banho para desgaseificar por 5 min.
- Submerja um rack de folhas de cobertura no copo de KOH e etanol e sonicate por 5 min.
- Transfira o rack de tampas do copo KOH/etanol para o copo somente etanol. Mergulhe o acumular para cima e para baixo no copo até que não haja miçangas.
NOTA: Tome cuidado para não perturbar as tampas ou soltar com força o rack no béquer. Isso fará com que as tampas saiam do rack ou causem respingos químicos. - Transfira cuidadosamente o rack de tampas do copo de etanol para um copo de água, mergulhando para cima e para baixo até que não haja miçangas.
- Submerja o rack de tampas no copo de água que ainda não foi usado e sonicate novamente por 5 min.
- Use uma garrafa para pulverizar o rack de folhas de cobertura com água até que ele escorra para fora das tampas suavemente. Repita com o etanol.
- Colocar as estantes a secar no forno a 90 °C durante 20 min. Armazene as prateleiras de coberturas gravadas à temperatura ambiente em recipientes fechados para evitar a contaminação antes do uso.
2. Polimerização do filamento de actina
- Fazer a solução T
- Em um tubo cônico de 50 mL, adicione 3,94 g de Tris-HCl e 0,147 g de CaCl2. Adicione a água RO para fazer um volume total de 50 mL e misture bem.
NOTA: As concentrações finais da Solução T são 500 mM Tris-HCl e 20 mM CaCl2, respectivamente. - Rotular o tubo Solução T e guardá-lo a 4 °C.
- Em um tubo cônico de 50 mL, adicione 3,94 g de Tris-HCl e 0,147 g de CaCl2. Adicione a água RO para fazer um volume total de 50 mL e misture bem.
- Criar buffer de TC
- Misture 40 mL de água RO e 1,5 mL de Solução T em um tubo cônico de 50 mL. Altere o pH para 8,0 adicionando pequenas quantidades de KOH concentrado. Adicione água para produzir 50 mL da solução e verifique o pH. Ajuste o pH, se necessário.
NOTA: O tampão TC final contém 5 mM Tris-HCl e 0,2 mM CaCl2 a pH 8. - Rotular o tubo TC e guardá-lo a 4 °C.
- Misture 40 mL de água RO e 1,5 mL de Solução T em um tubo cônico de 50 mL. Altere o pH para 8,0 adicionando pequenas quantidades de KOH concentrado. Adicione água para produzir 50 mL da solução e verifique o pH. Ajuste o pH, se necessário.
- Criar buffer FC
- Adicione 85 mL de água de RO, 10 mL de Solução T, 3,73 g de KCl e 0,041 g de MgCl2 a um frasco tampão de 100 mL. Modifique o pH para 7,5 adicionando pequenos volumes de KOH concentrado. Adicione água para fazer um volume final de 100 mL e verifique o pH.
NOTA: O tampão FC final contém 500 mM Tris-HCl, 500 mM KCl, 2 mM MgCl 2 e 2 mM CaCl2 a pH 7,5. - Rotular o tubo FC e guardá-lo a 4 °C.
- Adicione 85 mL de água de RO, 10 mL de Solução T, 3,73 g de KCl e 0,041 g de MgCl2 a um frasco tampão de 100 mL. Modifique o pH para 7,5 adicionando pequenos volumes de KOH concentrado. Adicione água para fazer um volume final de 100 mL e verifique o pH.
- Preparar o buffer geral de actina (GAB).
- Misture 485 μL de tampão TC, 10 μL de ATP de 10 mM e 5 μL de TDT de 50 mM em um tubo de microcentrífuga.
NOTA: As condições finais do buffer são Tris-HCl de 5 mM, CaCl 2 de0,2 mM, TDT de 0,5 mM e ATP de 0,2 mM. - Rotulá-lo como GAB e armazená-lo a 4 °C.
- Misture 485 μL de tampão TC, 10 μL de ATP de 10 mM e 5 μL de TDT de 50 mM em um tubo de microcentrífuga.
- Preparar tampão de polimerização de actina (APB).
- Misture 455 μL de tampão FC, 25 μL de ATP de 100 mM e 20 μL de TDT de 50 mM em um tubo de microcentrífuga.
NOTA: As condições finais do buffer são 50 mM Tris-HCl, 500 mM KCl, 2 mM MgCl 2, 2 mM CaCl 22 mM DTT e 5 mM ATP. - Rotular o tubo como APB e guardá-lo a 4 °C.
- Misture 455 μL de tampão FC, 25 μL de ATP de 100 mM e 20 μL de TDT de 50 mM em um tubo de microcentrífuga.
- Reconstituir actina
- Reconstituir a actina do músculo esquelético do coelho adicionando 100 μL de água deionizada a um frasco de 1 mg de actina liofilizada. Misture bem pipetando suavemente para cima e para baixo. Aliquotar em amostras de 5 μL, congelar rapidamente e armazenar as alíquotas de actina de 10 mg/mL a -80 °C.
- Reconstituir actina do músculo esquelético de coelho biotinilado adicionando 20 μL de água RO. Aliquotar em amostras de 5 μL, congelar rapidamente e armazenar as alíquotas de actina biotinilada de 1 mg/mL a -80 °C.
- Polimerização de actina não marcada com estabilização de faloidina rodamina
- Descongele um frasco para injetáveis de 10 mg/ml de actina e mantenha-o no gelo.
- Prepare o tampão GAB fresco, adicione 100 μL de GAB à alíquota da actina e misture suavemente pipetando para cima e para baixo. Incubar a solução no gelo por 1 h.
- Prepare APB fresco durante a incubação. Após incubação, polimerizar a actina em filamentos, adicionando 11 μL de APB à solução de actina. Misture bem pipetando suavemente para cima e para baixo. Coloque no gelo por 20 min.
- Adicionar 5 μL de faloidina marcada com rodamina à solução de filamento de actina recém-polimerizada. Deixe no gelo no escuro por 1 h.
- Conservar o frasco para injetáveis de actina de rodamina envolto em folha de alumínio no escuro a 4 °C.
NOTA: Sugere-se a utilização destes filamentos por um período máximo de 1 semana. A qualidade da FA pode ser confirmada todos os dias através de uma imagem rápida de uma célula de fluxo contendo apenas FAs e visualizando filamentos consistentes no dia a dia.
- Polimerização de actina biotinilada com estabilização de faloidina Alexa Fluor 488
- Descongele um frasco para injetáveis de 10 mg/ml de actina e 1 frasco para injetáveis de 1 mg/ml de actina biotinilada e mantenha-os no gelo.
- Faça um novo buffer GAB.
- Combinar os dois frascos para injetáveis (passo 2.8.1) numa relação actina:actina biotinilada de 10:1. Adicione 100 μL de GAB à mistura de actina e misture bem pipetando suavemente para cima e para baixo. Incubar no gelo por 1 h.
- Faça APB fresco durante a incubação.
- Após a etapa de incubação, polimerizar a actina adicionando 11 μL de APB à solução de actina. Misture bem pipetando para cima e para baixo suavemente. Incubar no gelo por 20 min.
- Adicione 5 μL de faloidina marcada com Alexa Fluor 488 e incube no gelo no escuro por 1 h.
- Conservar o frasco para injetáveis de actina biotinilado envolto em folha de alumínio no escuro a 4 °C.
NOTA: Estes filamentos podem ser utilizados por um período máximo de 1 semana.
3. Preparação de miosina e grânulos
- Reconstituir a Miosina II
- Gire brevemente para baixo (~ 5 s) miosina esquelética liofilizada II para coletá-la no fundo do tubo usando uma minicentrífuga padrão.
- Reconstituir a miosina para 10 mg/mL adicionando 100 μL de TDT 1 mM preparado em água RO.
- Diluir a solução de miosina de estoque 10x adicionando 10 μL de 10 mg/mL de miosina a 90 μL de TDT de 1 mM em água RO. Faça alíquotas de pequeno volume (1-5 μL), congele rapidamente e armazene a -80 °C.
NOTA: A atividade da miosina pode ser confirmada através da realização de um ensaio de filamento deslizante padrão, conforme publicado anteriormente46,47. Consulte a discussão para uma breve descrição.
- Limpeza de grânulos revestidos com estreptavidina
- Diluir 20 μL de 1 μm de esferas de estreptavidina em 80 μL de água RO. Lavar quatro vezes girando a 9.600 × g e reconstituindo em 100 μL de água RO.
- Sonicate durante 2 min a 40% de amplitude e conservar os grânulos lavados num rotador a 4 °C.
4. Preparação da célula de fluxo
- Prepare uma solução de poli-l-lisina (PLL) adicionando 30 mL de etanol a 100% a um tubo de 50 mL e adicionando 200 μL de poli-l-lisina a 0,1% p/v em água e misture bem.
- Adicione uma tampa gravada à solução PLL e deixe-a de molho por 15 minutos. Remova a tampa com uma pinça, tomando o cuidado de tocar apenas na borda da tampa quando ela for puxada para cima do tubo (ver Figura 1A-C). Agarre as tampas pelas bordas com uma mão enluvada.
- Seque a tampa com uma companhia aérea filtrada até que não haja etanol e nenhum resíduo na tampa.
- Aplique dois pedaços de fita adesiva de dupla face no meio de uma lâmina de microscópio, com 3-4 mm de distância um do outro. Rasgue ou corte o excesso de fita que fica pendurado na borda do slide.
- Adicione a tampa revestida de PLL em cima da fita perpendicular ao longo eixo da lâmina do microscópio (formando um T) para formar um canal.
- Use um tubo pequeno para comprimir a tampa na fita e o microscópio deslize completamente até que a fita fique transparente (Figura 1A). Certifique-se de que não há bolhas na fita, pois isso pode causar vazamento do canal de fluxo.
NOTA: A célula de fluxo pode conter um volume de 10-15 μL.
5. Preparação do feixe de actomiosina
- Em tubos separados, diluir cada tipo de filamento de actina (marcado com rodamina e biotinilado 488) 600x misturando 0,5 μL da respectiva actina marcada com 300 μL de APB. Adicione mais 5 μL da faloidina correspondentemente rotulada a cada tubo e incube no gelo no escuro por 15 min.
- À solução de actina biotinilada, adicionar um sistema de eliminação de oxigênio de 1 μL de beta-D-glicose a 500 mg/mL, 1 μL de glicose oxidase a 25 mg/mL e 1 μL de catalase a 500 unidades/mL. Adicionar 1 μL de 100 mM de ATP e 1 μL de grânulos de estreptavidina 100x diluídos e limpos. Mexa suavemente com uma ponta de pipeta. Coloque a suspensão num rotador a 4 °C enquanto o resto do feixe de actomiosina está a ser montado.
- Adicionar 15 μL da actina de rodamina diluída à célula de fluxo PLL (Figura 1D). Wick o excesso de solução através da célula de fluxo, mas não permita que o canal de fluxo fique seco. Incubar por 10 min em uma câmara de umidade.
NOTA: As câmaras de umidade podem ser feitas de caixas de ponta de pipeta vazias com água adicionada ao fundo e a tampa coberta de folha de alumínio para bloquear a luz. - Preparar uma solução de caseína de 1 mg/ml em APB.
- Adicionar 15 μL de 1 mg/mL de caseína para evitar a ligação inespecífica dos componentes subsequentes (Figura 1E). Incubar por 5 min em uma câmara de umidade.
- Adicionar a concentração desejada de miosina à suspensão de actina e grânulos biotinilados a partir da fase 5.2. Mexa suavemente com a ponta da pipeta e, em seguida, adicione imediatamente 15 μL da suspensão do passo 5.2 + a concentração de miosina desejada à célula de fluxo (Figura 1F,G). Incubar por 20 min. Sele as extremidades abertas da célula de fluxo com esmalte para evitar a evaporação durante os experimentos de imagem e aprisionamento óptico.
NOTA: Uma concentração de solução de miosina de 1 μM produz um empacotamento robusto e pode ser usada como ponto de partida para a personalização desejada do ensaio (ver Figura 2).
6. Medições de força usando armadilha óptica (NT2 Nanotracker2)
NOTA: Embora o protocolo abaixo seja especificamente para o sistema NT2, este ensaio pode ser usado com outros sistemas de interceptação óptica, incluindo aqueles que são personalizados, que também têm recursos de fluorescência. O fluxo de trabalho geral permanece o mesmo de colocar a superfície do slide em foco, realizar calibrações de contas e adquirir dados encontrando feixes de actina fluorescentes. Para o sistema NT2, a Figura Suplementar S1, a Figura Suplementar S2, a Figura Suplementar S3, a Figura Suplementar S4, a Figura Suplementar S5, a Figura Suplementar S6 e a Figura Suplementar S7 fornecem detalhes do sistema de armadilhagem óptica e da interface do software.
- Ligue a caixa de controle e o laser (Figura suplementar S1).
- Inicie o software de computador de armadilha óptica clicando no ícone JPK Nanotracker na área de trabalho.
- Acorde o controle remoto clicando no botão Logitech no centro (Figura Suplementar S2).
- Ligue o módulo de fluorescência alternando o interruptor liga/desliga (Figura S3 Suplementar).
- Gire a torre do cubo de filtro para obter imagens de campo brilhante (Figura suplementar S4).
- Quando o sistema estiver pronto, ligue o laser usando o botão Laser Power no canto inferior esquerdo da tela a 50 mW e deixe-o estabilizar por 30 min (Figura suplementar S5).
- Clique sequencialmente nos botões Iluminação, Câmera, Objetivo e Movimento do Palco dentro do software para abrir essas janelas para visualização e manipulação durante o experimento. Ligue a iluminação do microscópio clicando no botão Ligar/Desligar e definindo-a para a potência máxima clicando e arrastando a barra até a direita (Figura Suplementar S5).
- Abra a área da amostra e remova o suporte da amostra do estágio do microscópio. Adicione a célula de fluxo, prenda-a com os suportes de amostra de metal e certifique-se de que a lâmina com a tampa esteja na parte inferior.
- Adicione 30 μL de água RO ao centro da objetiva inferior. Não deixe a ponta da pipeta tocar na lente. Reinsira o estágio de amostra.
NOTA: Como o sistema NT2 usa uma objetiva de imersão em água como objetivo de aprisionamento, a mídia de imersão pode ser diferente dependendo da objetiva de aprisionamento na configuração do usuário. - Levante a objetiva inferior usando as setas de controle na tela ou L2 no controle remoto até que o talão de água toque a tampa (Figura Suplementar S5).
- Abaixe a objetiva superior até que cerca de metade da distância até a célula de fluxo seja alcançada usando as setas na tela ou R2 no controle remoto. Adicione 170 μL de água RO ao topo da célula de fluxo diretamente abaixo da objetiva superior. Abaixe a objetiva superior até que ela quebre a tensão superficial da água e forme um menisco.
- Mova o estágio do microscópio usando a almofada de seta no controle remoto até que a borda da fita adjacente ao canal de fluxo seja alcançada. Feche a porta da amostra.
NOTA: Um "clique" ao fechar a porta da amostra indica que o obturador a laser está agora aberto. Este é um recurso de segurança que só permite que o obturador abra se a porta estiver fechada. - Usando a janela Objetivo na tela, coloque a borda da fita em foco trazendo a objetiva inferior chamada Objetivo a Laser para cima clicando na seta superior usando os controles na tela. Faça o mesmo para o objetivo superior clicando na seta inferior (Figura Suplementar S5).
NOTA: As setas duplas movem o objetivo ou o estágio mais rapidamente. A borda da fita é usada para focar porque é um objeto grande e fácil de encontrar que está perto da superfície da tampa. Bolhas de ar dentro da fita são outra opção. No entanto, isso não é necessário se o usuário tiver uma rotina automatizada para encontrar o foco da superfície ou um método interno preferido. - Quando a fita estiver em foco, feche parcialmente a íris na parte superior do purgador óptico. Traga a objetiva superior para baixo até que a forma de polígono da íris seja visível. Coloque essas bordas em foco, reabra a íris e acople os objetivos usando clicando no ícone de cadeado (Figura Suplementar S5).
- Encontre um grânulo flutuante e prenda-o clicando no botão Trap Shutter , que abrirá o obturador e permitirá que o laser de armadilha atinja a amostra. Clique no cursor Trap na tela e arraste-o para mover a localização do laser de trapping . Uma vez presa, calibre o talão para correlacionar as medições de tensão à força e ao deslocamento.
- Clique no botão Calibração . Ajuste a rotina de calibração com base na análise de espectros de potência e ajuste a frequência de canto dentro do software para as direções X, Y e Z (Figura S6 Suplementar).
- Clique em Configurações. Digite o diâmetro do talão (1.000 nm) e digite a temperatura do estágio encontrada no canto inferior esquerdo da janela do software. (ver Figura suplementar S6).
- Clique em Armadilha 1. Clique em X Signal. Clique em Executar para executar o ajuste de frequência de canto. Clique e arraste dentro da janela para otimizar o ajuste da função. Clique em Usá-lo para valores de sensibilidade e rigidez. Clique em Aceitar Valores. Repita para os sinais Y e Z. Feche a janela. (ver Figura suplementar S6).
NOTA: Rotinas de calibração de talões em outros sistemas de aprisionamento óptico ou sistemas personalizados que foram testados de forma robusta pelo usuário, como o método de equipartição ou o método de força de arrasto, também são aceitáveis57,58. - Encontre um feixe de actomiosina procurando por contas ligadas a FAs na superfície da tampa.
- Quando uma conta não lotada por outras contas flutuantes for detectada, observe os FAs ao seu redor por imagem de fluorescência para verificar a presença de um feixe.
- Verifique se um feixe está presente procurando por ambos os FAs fluorescentes colocalizados. Ligue a fonte de luz branca e use o cubo de filtro apropriado para obter a imagem de cada filamento de actina girando a torre (cubos de filtro de excitação de 488 nm e 532 nm para Alexa Fluor 488 e excitação de rodamina, respectivamente). Consulte a Figura suplementar S4.
NOTA: Um experimento de controle para verificar a intensidade de fluorescência de AFs únicos pode ser útil na identificação de feixes que são compostos de um único filamento marcado com rodamina única e 488, ou aplicável a qualquer conjunto de fluoróforos que o usuário escolha usar. - Uma vez verificado, prenda o talão preso ao filamento superior do feixe clicando no botão Trap Shutter .
- Use os controles na tela para registrar os dados clicando no botão Osciloscópio (Figura Suplementar S7). Para visualizar as medições sem registrar os dados, clique em Iniciar. Para salvar todos os dados, clique em Salvar automaticamente. Para registrar as medições, clique em Iniciar registro. Escolha quais dados devem ser visualizados em tempo real (posição, força, direção x, direção y) escolhendo no menu suspenso sinal X ou sinal Y. Lembre-se de que a direção x é da esquerda para a direita e a direção y é para cima e para baixo na tela. Consulte a Figura suplementar S7.
NOTA: Os dados serão salvos como arquivos .out e incluem tempo, tensão, deslocamento e força para cada direção. Esses arquivos podem ser exportados para outros softwares para visualização e análise.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
As células de fluxo que contêm os sistemas de feixe de actomiosina são de um projeto padrão, consistindo de uma lâmina de microscópio e uma lâmina de cobertura gravada separada por um canal feito de fita adesiva de dupla face (Figura 1). O ensaio é então construído a partir do deslizamento de cobertura usando introduções em estágios, conforme descrito no protocolo. O ensaio final consiste em filamentos de actina marcados com rodamina; a concentração de miosina desejada (1 μM foi utilizado para os resultados representativos na Figura 2 e Figura 3); filamentos de actina biotinilados, Alexa Fluor 488; 1 μm de contas de estreptavidina; o sistema de eliminação de oxigénio; ATP; e buffer APB. Múltiplos feixes serão formados por célula de fluxo, e as concentrações de actina descritas acima fornecem espaçamento adequado entre os feixes para garantir que não haja interações indesejadas. Isso também facilita a obtenção de múltiplas medições de força por célula de fluxo para aumentar a eficiência da aquisição de dados. Os perfis de força devem ser reprodutíveis dentro de uma célula de fluxo e de célula de fluxo para célula de fluxo.
Embora o protocolo acima seja voltado para o uso de uma configuração comercial de aprisionamento óptico, a célula de fluxo e o ensaio apresentados aqui poderiam ser facilmente utilizados para um instrumento comercial diferente ou uma configuração de aprisionamento óptico personalizada, juntamente com um microscópio ou estágio de microscópio e possuindo recursos de imagem de fluorescência. Uma vez que todas as adições de células de fluxo estejam completas de acordo com o protocolo acima, os feixes de actomiosina na lâmina (Figura 1) estão prontos para medição imediata. A célula de fluxo é adicionada ao estágio de microscópio de armadilha óptica, várias medições de calibração de contas são adquiridas e os feixes são identificados através da colocalização por fluorescência dos filamentos do feixe. Um talão ligado a um feixe é preso e o deslocamento e a medição de força correspondente começam. O usuário pode observar a aquisição de dados em tempo real no monitor do computador. Dependendo da concentração de miosina usada na célula de fluxo, o feixe pode começar a exibir movimento substancial imediatamente, ou pode levar 30 s-1 min para efetivamente ver um aumento no deslocamento / força.
Um traço de força representativo é mostrado na Figura 3A , onde os motores de miosina exibem uma rampa constante em força, seguida por um platô. É típico ver esses tipos de vestígios se desenvolverem ao longo de 2-5 minutos. No entanto, também é possível medir feixes de actomiosina que não geram força líquida (Figura 3B). Estes vestígios aparecem como ruído de base ou não apresentam um aumento líquido substancial da força ao longo dos anos 90. Isto é provavelmente devido a uma baixa concentração local de motor que não permite o deslizamento produtivo, ou o feixe está em uma orientação paralela desfavorável, onde as extremidades mais e menos dos filamentos estão alinhadas.
Como o conteúdo da célula de fluxo pode ser suscetível à degradação do laser de iluminação e aprisionamento incidente, aquecimento local no deslizamento ao longo do tempo e geração de espécies radicais de oxigênio, é altamente recomendável não usar a mesma célula de fluxo por mais de 1 h. Para máxima eficiência, sugere-se ter outro ensaio incubando enquanto adquire dados. O rastreamento de deslocamento/força pode ser exportado do software de aprisionamento óptico para o Excel, Matlab, Igor ou outros programas de gerenciamento de dados para filtragem e análise adicionais. Os dados que podem ser extraídos de tais experimentos de conjunto de aprisionamento óptico / pacote incluem diferentes tipos de perfis de geração de força (linha de base, rampa/platô) sob diferentes condições de ensaio, velocidade de geração de força, geração de força máxima, cinética do conjunto e comportamento de passo através de tamanhos de passo e tempos de permanência entre etapas ou equipes de etapas, bem como taxa de serviço. O usuário também pode alterar as condições do ensaio para comparar como a adição de diferentes tipos de motores de miosina, a adição de proteínas de ligação à actina ou a alteração das condições do tampão influenciam essas características de geração de força do conjunto.
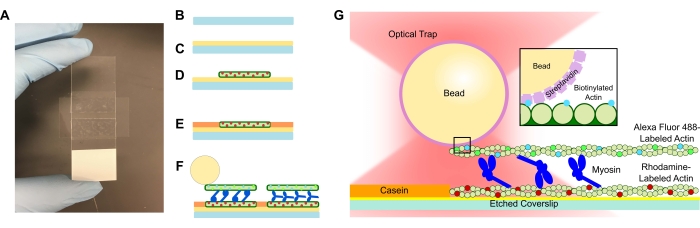
Figura 1: Esquema de ensaio. (A-C) As folhas de cobertura gravadas são revestidas em poli-L-lisina e usadas para formar a célula de fluxo usando fita dupla face e uma lâmina de microscópio. Introduções cronometradas e etapas de incubação descritas no protocolo resultam em actina estabilizada com faloidina marcada com rodamina como molde ou filamento inferior (D), seguida de bloqueio de caseína para evitar a ligação não específica (E) e (F) actina biotinilada estabilizada com faloidina Alexa Fluor 488 como carga ou filamento superior, e equipes de miosina II que deslizam os filamentos e geram força quando o ATP é introduzido. A geometria dos motores e a natureza da reticulação dentro do feixe podem variar sob diferentes condições, como a concentração de sal59. Estudos prévios demonstraram que o domínio cauda da miosina tem a capacidade de interagir com filamentos de actina e motilidade lenta do conjunto46. No entanto, cabeças de miosina em experimentos pesados de meromiosina demonstram a ligação de cada cabeça a filamentos de actina adjacentes60. (G) As esferas de estreptavidina são utilizadas como alça óptica para a armadilha e ligam-se exclusivamente ao filamento de actina biotinilado da carga, o que ajuda a validar que os feixes adequados são formados na lâmina. Por favor, clique aqui para ver uma versão maior desta figura.

Figura 2: Feixes de actomiosina fluorescentes. Quatro encontros diferentes de filamentos de actina e feixes dentro do ensaio de feixe apresentados na Figura 1. O filamento de actina biotinilado de carga superior com o canal de faloidina Alexa Fluor 488 é mostrado à esquerda, e o filamento de actina de molde inferior com o canal de faloidina de rodamina está à direita. Na parte inferior, a mesma figura é mostrada com linhas coloridas sobrepostas para ajudar a guiar o olho. (A) Um filamento de actina superior é encontrado perto de um filamento de actina inferior, mas tem uma sobreposição incompleta. Isso não seria usado para experimentos de pacote. (B) Os filamentos de actina superior e inferior são colocalizados, e a intensidade de cada filamento confirma que são filamentos individuais dentro do feixe. Isso seria um bom candidato para experimentos de pacote. (C) Um grande feixe de filamentos de rodamina auto-montados é encontrado no fundo. Embora exista um filamento de actina superior correspondente que é colocalizado, existem muitos filamentos inferiores presentes; portanto, não seria usado para experimentos de pacote. Este é também um exemplo de como quando múltiplos filamentos de actina do mesmo tipo são agrupados, a intensidade da fluorescência aumenta. O usuário pode utilizar isso como um medidor para julgar filamentos únicos versus feixes do mesmo tipo de filamento. (D) Um filamento inferior está presente sem filamento superior correspondente, confirmando também que não sangrou completamente. Isso não seria usado para experimentos de pacote. Notamos que a intensidade dos filamentos no canal Alexa Fluor 488 é baixa e acreditamos que seja devido ao conjunto de filtros que está sendo usado (Filter Set 09 da Zeiss). O conjunto de filtros usado para o canal de rodamina é o Conjunto de Filtros 43 da Zeiss. Por favor, clique aqui para ver uma versão maior desta figura.

Figura 3: Geração de força do conjunto de miosina II. Traços representativos de motores esqueléticos de miosina II gerando força dentro da hierarquia estrutural de actina in vitro construída. Os motores da miosina estão trabalhando juntos para gerar força coletiva e produtivamente até que um platô seja atingido e a força seja sustentada (A) ou experimente antagonização perto da linha de base (B). Por favor, clique aqui para ver uma versão maior desta figura.
Figura suplementar S1: Armadilha óptica Bruker/JPK Nanotracker2. (A) Monitor de computador. (B) Teclado de computador. (C) Torre de computador. (D) Caixa do controlador. (E) Fonte de alimentação a laser. (F) Caixa óptica de armadilha óptica. (G) Microscópio invertido. (H) Porta para o estágio do microscópio. (I) Controle deslizante do polarizador para alternar entre imagens de contraste de campo brilhante e interferência diferencial. Clique aqui para baixar este arquivo.
Figura suplementar S2: Controle remoto para armadilha óptica. (A) Teclado para posicionar o estágio motorizado. (B-C) Ajuste a posição do purgador. (D) A, X e B ligam e desligam o obturador principal, o obturador trap 1 e o obturador trap 2, respectivamente. (E) O botão Logitech é usado para ativar o controlador. (F) Os botões para cima e para baixo que são usados para posicionar o objetivo de interceptação. (G) Os botões para cima e para baixo que são usados para posicionar o objetivo de detecção. Observe que o controle remoto não é necessário, e todas essas manipulações podem ser realizadas no software. No entanto, é conveniente ser capaz de controlar os objetivos e a posição do estágio enquanto olha para o ambiente do estágio do microscópio. Clique aqui para baixar este arquivo.
Figura suplementar S3: Módulo de fluorescência para armadilha óptica. A fonte de luz branca de fluorescência 89North PhotoFluor é acoplada à parte de trás do microscópio invertido. Ele é ligado e desligado com um botão de alternância (seta). Clique aqui para baixar este arquivo.
Figura suplementar S4: Torreta de cubo de filtro de fluorescência. A torre (seta) pode ser girada para usar o cubo de filtro necessário para a imagem em corantes DIC, rodamina ou Alexa Fluor 488. Observe que os cubos de filtro podem ser alternados para personalizar a configuração para usar fluoróforos diferentes. Clique aqui para baixar este arquivo.
Figura suplementar S5: Software Nanotracker2. (A) Botão de alimentação e controle do laser. (B) Janela de posicionamento objetiva. As setas direcionais são usadas para mover os objetivos de detecção (superior) e trapping (inferior). Setas duplas movem os objetivos a uma velocidade maior. O botão azul e vermelho na parte inferior esquerda desacopla os objetivos e os retrai de volta à sua posição original. Isso é necessário para quando se coletam amostras dentro e fora do estágio do microscópio. O terceiro botão da esquerda com os objetivos e o ícone de cadeado "acopla" os objetivos para que, quando ambos estiverem em foco e alcançarem a iluminação Kohler, o usuário possa mover os objetivos de armadilha e detecção para cima e para baixo no eixo z. (C) Janela de posicionamento da amostra usada para mover o estágio do microscópio nos eixos x e y. Setas duplas movem o palco a uma velocidade maior. Esta janela é ativada clicando no ícone de seta para cima / para baixo e para a esquerda / direita no menu superior. (D) Janela de visualização da câmera. O ícone de chave inglesa pode ser usado para definir condições de imagem personalizadas. Esta janela é ativada clicando no ícone Câmera no menu superior. (E) Janela de iluminação do microscópio. Esta janela é ativada clicando no ícone Lâmpada no menu superior. Clique aqui para baixar este arquivo.
Figura suplementar S6: Janela de calibração. (A) Esta janela é usada para calibração de contas e é ativada clicando no ícone Cal no menu superior. Para calibrar um talão, um melhor ajuste da frequência de canto é realizado nos sinais x, y e z. (B) Para cada sinal, escolha o botão de sinal apropriado no canto superior esquerdo. (C) Clique em executar e otimize o ajuste clicando e arrastando dentro da janela verde (D). (E) Uma vez satisfeito com o ajuste, clique em Usá-lo para sensibilidade e rigidez. Isso permitirá registrar o deslocamento em nanômetros e a força em piconewtons. (F) Em seguida, clique em Aceitar Valores no canto inferior esquerdo. Repita para as direções y e z. Clique aqui para baixar este arquivo.
Figura suplementar S7: Janela Aquisição de dados. Esta janela é usada para adquirir dados de posição e força e permite que o usuário veja as medições em tempo real. (A) Esta janela é ativada clicando no ícone x,t no menu superior. (B) O usuário pode alternar entre a visualização dos sinais x e y. (C) Clique em Iniciar para começar a visualizar os dados. Clique em Salvar automaticamente para salvar os dados. Clique em Iniciar Registro para começar a gravar e salvar dados. Clique aqui para baixar este arquivo.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Um estudo in vitro utilizando pinças ópticas combinadas com imagens de fluorescência foi realizado para investigar a dinâmica de conjuntos de miosina interagindo com filamentos de actina. Os feixes de actina-miosina-actina foram montados usando miosina muscular II, actina de rodamina na parte inferior do feixe e na superfície da lâmina de cobertura e filamentos de actina biotinilados marcados com 488 no topo do feixe. A proteína actina do músculo coelho foi polimerizada e estabilizada usando tampões gerais de actina (GAB) e tampões polimerizantes de actina (APB). GAB e APB devem ser preparados recentemente todos os dias no laboratório usando ATP, buffer FC e buffer TC. A miosina II muscular foi utilizada para formar os sanduíches de actina-miosina-actina. A faloidina foi utilizada para coloração fluorescente dos filamentos de actina, bem como estabilização in vitro.
A atividade da miosina pode ser confirmada através da realização de um ensaio de filamento deslizante padrão, conforme publicado anteriormente46,47. A miosina II e seus subfragmentos podem se ligar à superfície da cobertura em uma variedade de orientações, e a presença do domínio da cauda pode retardar o deslizamento do filamento em comparação com ensaios usando meromiosina pesada46,48,49. No entanto, o deslizamento e o movimento da superfície ainda podem ser observados. Uma demonstração mais aparente da atividade da miosina é a quebra ativa do filamento de actina que pode ser observada onde filamentos de actina mais longos são quebrados em fragmentos menores que então deslizam em várias direções. Isso ocorre devido à alta concentração de motores ativos na superfície, tem sido observada por múltiplos laboratórios, e não ocorre sem a presença de motores ativos de miosina 42,50,51,52,53,54. Além disso, o ensaio de feixe aqui apresentado auxilia no alívio de problemas de motilidade que têm sido associados principalmente ao ensaio de filamento deslizante, como a variedade de orientações de ligação do motor em uma tampa de vidro, porque o ensaio de feixe envolve o bloqueio de caseína da superfície de vidro para que os motores se liguem dentro do feixe 47,55,56.
O primeiro passo é adicionar filamentos de actina de rodamina como filamento inferior ou molde a uma tampa revestida de poli-L-lisina em uma célula de fluxo. A poli-L-lisina é utilizada para promover a ligação da actina, uma vez que a polilisina é carregada positivamente, enquanto a actina tem cargas negativas e tem sido utilizada em preparações de ensaios in vitro citoesqueléticos anteriores61,62,63. Antes da formação do feixe, diferentes diluições de actina foram adicionadas a uma célula de fluxo para otimizar a concentração de actina. Neste caso, 600x do estoque foi a diluição ideal que produziu um número suficiente de filamentos molde para a formação do feixe, mas com espaçamento adequado para que os feixes fossem individualizados. A diluição foi realizada utilizando-se o tampão APB. A adição de actina de rodamina foi seguida por uma camada de caseína para bloquear a superfície e evitar a ligação não específica. A célula de fluxo foi incubada por 30 min e lavada após incubação com tampão para lavar quaisquer filamentos de actina não ligados. Finalmente, uma combinação de miosina, 488/actina de biotina e contas revestidas com estreptavidina foram adicionadas à célula de fluxo para facilitar a formação do feixe de actina-miosina. A concentração do talão deve ser tal que haja o suficiente para ligar feixes ligados à superfície e suficiente em suspensão para facilitar a calibração. No entanto, uma concentração muito alta de contas pode causar dificuldade durante os experimentos de aprisionamento devido a contas vizinhas caindo na armadilha a laser e interrompendo a medição. Os motores de miosina são adicionados à combinação antes de injetá-la na lâmina para que os motores de miosina não se agreguem preventivamente com a carga ou o filamento de actina biotinilado superior e, assim, liguem a rodamina inferior para agrupar filamentos de actina biotinilados.
O sistema de aprisionamento óptico NT2 é um purgador óptico comercial com campo de brilho combinado, contraste de interferência diferencial (DIC) e modalidades de imagem de epifluorescência. Ele é acoplado a um microscópio invertido Zeiss AxioObserver 3 com objetivos de aprisionamento e detecção de imersão em água 100x/NA 1.46 e 63x/NA 1.0. O sistema é equipado com capacidade de retenção de clique e arrasto de uma armadilha a laser e pode ser usado durante a imagem em qualquer uma das modalidades listadas anteriormente. Os feixes formados são detectados e confirmados usando imagens de fluorescência. Ter uma fonte de luz branca com cubos de filtro apropriados (GFP / FITC e TRITC / CY3) permite uma comutação rápida entre imagens de filamento. As FAs colocalizadas foram verificadas visualizando-se as FAs nos diferentes comprimentos de onda de excitação antes de realizar cada medição de força usando pinças ópticas. Como os filamentos podem fotobranquear rapidamente, mesmo com um reagente de eliminação de oxigênio, sugere-se que os pesquisadores otimizem os parâmetros de visualização, como intensidade e tempo de exposição, antes de realizar os experimentos do feixe.
O aprisionamento óptico foi empregado para a realização das medidas de força, utilizando-se os grânulos de estreptavidina na presença de ATP para ligar o filamento de actina de carga biotinilado e ativar a geração de força da miosina como transdutor de força. Os dados de deslocamento e força versus tempo obtidos por aprisionamento óptico foram extraídos do software de aprisionamento para análise. No entanto, o software de armadilhagem comercial também fornece rotinas de análise que podem ser utilizadas, ou algoritmos personalizados em outros programas podem ser programados pelo usuário para visualizar e analisar dados de armadilhagem. Em sistemas de retenção óptica personalizados, o usuário pode ter lasers de excitação em vez de uma fonte de luz branca com filtros, que também são aceitáveis de usar. Além disso, os corantes fluorescentes podem ser alterados para serem adequados ao equipamento existente que um usuário pode ter se os espectros de emissão não se sobreporem e causarem sangramento.
Notamos que o ensaio apresentado é um ensaio de linha de base que pode ser posteriormente personalizado pelo usuário, dependendo de sua questão de pesquisa dentro do domínio da mecânica do conjunto de actomiosina. O fluxo de trabalho geral também pode ser aplicado a outros sistemas de conjunto citoesquelético in vitro que possam ser de interesse, como ensaios de feixes de microtúbulos que formam modelos mínimos de fuso mitótico 32,61,63,64,65,66. As modificações podem incluir, mas não estão limitadas a alterar os rótulos de fluoróforo que são adequados à configuração existente do usuário; alterar a concentração, o construto ou o isotipo da miosina; e condições de titulação do amortecedor, entre outros aspectos.
Desafios potenciais são possíveis ao realizar este ensaio. Ao formar os feixes de actina-miosina, a concentração de miosina dentro dos feixes de actina pode não ser homogênea em toda a lâmina. Para acomodar isso, vários feixes em toda a lâmina serão medidos para garantir que a distribuição do motor e os perfis de geração de força sejam adequadamente amostrados. Também é um desafio saber a orientação do feixe se isso for necessário para a interpretação dos dados de força. Assim, vários ensaios devem ser tomados para cada pacote. Pode-se também incorporar a marcação da extremidade do filamento de actina através de gelsolina fluorescente ou contas revestidas de gelsolina de um tamanho menor do que a alça de aprisionamento óptico. A imagem de fluorescência também pode ser usada para observar as forças dos componentes x e y para deduzir a orientação do feixe. Além disso, como o estado de agregação da miosina é altamente influenciado pela força iônica do tampão com a formação de filamentos espessos ocorrendo após a rápida diluição do KCl, a concentração de sal tampão deve ser monitorada adequadamente67,68.
Estudos anteriores que usaram outros métodos in vitro, como ensaios de deslizamento, foram úteis para identificar o papel dos domínios da miosina e estudar a configuração e as interações entre a miosina e outras proteínas de ligação à actina. No entanto, esses métodos têm uma desvantagem na medida em que a ligação da miosina em uma superfície rígida limitará o potencial de coordenação entre os motores de miosina e, portanto, o feedback mecanosense que ocorre para determinar se o conjunto motor está em um modo de relação de serviço alto ou baixo 33,35,41,69. Além disso, o aprisionamento óptico com redes motoras de miosina única não fornece uma compreensão clara de como os motores de miosina interagem uns com os outros e com os filamentos de actina. O protocolo aqui desenvolvido permite a investigação da dinâmica do conjunto motor da miosina dentro de uma rede de actina hierárquica e compatível. Também é personalizável em termos de características do conjunto motor-filamento, como concentração, isoforma e ambiente tampão, entre outros aspectos, para permitir a investigação sistemática. O protocolo apresentado é uma plataforma para estudos futuros de redes de actomiosina mais complexas e mantém a precisão das medições de deslocamento e geração de força facilitadas pelo aprisionamento óptico que tradicionalmente tem sido usado para estudos de molécula única.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores não têm conflitos de interesse a declarar.
Acknowledgments
Este trabalho é apoiado em parte pela University of Mississippi Graduate Student Council Research Fellowship (OA), pela University of Mississippi Sally McDonnell-Barksdale Honors College (JCW, JER), pelo Mississippi Space Grant Consortium sob o número de concessão NNX15AH78H (JCW, DNR) e pela American Heart Association sob o número de concessão 848586 (DNR).
Materials
| Name | Company | Catalog Number | Comments |
| Actin protein (biotin): skeletal muscle | Cytoskeleton | AB07-A | Biotinylated actin protein |
| Actin protein, rabbit skeletal muscle | Cytoskeleton | AKL99-A | Actin protein |
| Alexa Fluor 488 Phalloidin | Invitrogen | A12379 | Actin stabilizer and Alexa Fluor 488 stain |
| ATP | Fisher scientific | BP413-25 | Required for actin assembly and myosin motility |
| Beta-D-glucose | Fisher scientific | MP218069110 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Blotting Grade Blocker (casein) | Biorad | 1706404 | Used to block surface from non-specific binding |
| CaCl2 | Fisher scientific | C79500 | Calcium chloride, provides the necessary control over the dynamics of actin myosin network |
| Catalase | Fisher scientific | ICN10040280 | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| Coverslips | Fisher scientific | 12544C | Used to make flow cells |
| DTT | Fisher scientific | AC327190010 | Used for buffer preparation |
| Ethanol | Fisher scientific | A4094 | Regent used for cleaning coverslips |
| Glucose oxidase | Fisher scientific | 34-538-610KU | Part of oxygen scavenging system used to reduce photobleaching during fluorescence imaging |
| KCl | Fisher scientific | P217-500 | Used for buffer preparation |
| KOH | Fisher scientific | P250-1 | Used to etch coverslips and adjust buffer pH |
| MgCl2 | Fisher scientific | M33-500 | Used for buffer preparation |
| Microscope slides | Fisher scientific | 12-544-2 | Used to make flow cells |
| Myosin II protein: rabbit skeletal muscle | Cytoskeleton | MY02 | Full length myosin motor protein isolated from rabbit skeletal muscle |
| Nanotracker2 | Bruker/JPK | NT2 | Optical trapping instrument |
| Poly-l-lysine | Sigma-Aldrich | P8920 | Facilities adhesion of actin filaments onto glass surface of the coverslip |
| Rhodamine Phalloidin | Cytoskeleton | PHDR1 | Actin stabilizer and rhodamine fluorescent stain |
| Streptavidin beads, 1 μm | Spherotech | SVP-10-5 | Optical trapping handle |
| Tris-HCl | Fisher scientific | PR H5121 | Used for buffer preparation |
References
- Goldstein, L. S. Kinesin molecular motors: transport pathways, receptors, and human disease. Proceedings of the National Academy of Sciences of the United States of America. 98 (13), 6999-7003 (2001).
- Lee Sweeney, H., Holzbaur, E. L. F.
- O'Connell, C. B., Tyska, M. J., Mooseker, M. S. Myosin at work: Motor adaptations for a variety of cellular functions. Biochimica et Biophysica Acta - Molecular Cell Research. 1773 (5), 615-630 (2007).
- Kaya, M., Tani, Y., Washio, T., Hisada, T., Higuchi, H. Coordinated force generation of skeletal myosins in myofilaments through motor coupling. Nature Communications. 8, 1-13 (2017).
- Akhshi, T. K., Wernike, D., Piekny, A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton. 71 (1), 1-23 (2014).
- Brawley, C. M., Rock, R. S. Unconventional myosin traffic in cells reveals a selective actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America. 106 (24), 9685-9690 (2009).
- Hartman, M. A., Spudich, J. A. The myosin superfamily at a glance. Journal of Cell Science. 125 (7), 1627-1632 (2012).
- Spudich, J. A., et al.
- Sommese, R. F., et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proceedings of the National Academy of Sciences of the United States of America. 110 (31), 12607-12612 (2013).
- Nag, S., et al. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nature Structural & Molecular Biology. 24 (6), 525-533 (2017).
- Kawana, M., Sarkar, S. S., Sutton, S., Ruppel, K. M., Spudich, J. A. Biophysical properties of human b-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Science Advances. 3 (2), 1-11 (2017).
- Girolami, F., et al. Novel α-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: A massively parallel sequencing study. Circulation: Cardiovascular Genetics. 7 (6), 741-750 (2014).
- Debold, E. P., et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. American Journal of Physiology - Heart and Circulatory Physiology. 293 (1), 284-291 (2007).
- Barron, J. T.
- Duke, T. A. J.
- Vilfan, A., Duke, T.
- Huxley, A. F. Muscle structure and theories of contraction. Progress in Biophysics and Biophysical Chemistry. 7, 255-318 (1957).
- Huxley, H. E. Fifty years of muscle and the sliding filament hypothesis. European Journal of Biochemistry. 271 (8), 1403-1415 (2004).
- Kad, N. M., Kim, S., Warshaw, D. M., VanBuren, P., Baker, J. E. Single-myosin crossbridge interactions with actin filaments regulated by troponin-tropomyosin. Proceedings of the National Academy of Sciences of the United States of America. 102 (47), 16990-16995 (2005).
- Veigel, C., Molloy, J. E., Schmitz, S., Kendrick-Jones, J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nature Cell Biology. 5 (11), 980-986 (2003).
- Spudich, J. A.
- Simmons, R. M., Finer, J. T., Chu, S., Spudich, J. A. Quantitative measurements of force and displacement using an optical trap. Biophysical Journal. 70 (4), 1813-1822 (1996).
- Finer, J. T., Simmons, R. M., Spudich, J. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 368 (6467), 113-119 (1994).
- Kron, S. J., Uyeda, T. Q. P., Warrick, H. M., Spudich, J. A. An approach to reconstituting motility of single myosin molecules. Journal of Cell Science. 98, 129-133 (1991).
- Molloy, J. E., Burns, J. E., Kendrick-Jones, B., Tregear, R. T., White, D. C. S. Movement and force produced by a single myosin head. Nature. 378 (6553), 209-212 (1995).
- Ruegg, C., et al. Molecular motors: Force and movement generated by single Myosin II molecules. Physiology. 17 (5), 213-218 (2002).
- Nayak, A., et al. Single-molecule analysis reveals that regulatory light chains fine-tune skeletal myosin II function. Journal of Biological Chemistry. 295 (20), 7046-7059 (2020).
- Dupuis, D. E., Guilford, W. H., Wu, J., Warshaw, D. M.
- Tyska, M. J., et al. Two heads of myosin are better than one for generating force and motion. Proceedings of the National Academy of Sciences of the United States of America. 96 (8), 4402-4407 (1999).
- Tyska, M. J., Warshaw, D. M.
- Finer, J. T., et al.
- Al Azzam, O., Trussell, C. L., Reinemann, D. N. Measuring force generation within reconstituted microtubule bundle assemblies using optical tweezers. Cytoskeleton. 78 (3), 111-125 (2021).
- Wagoner, J. A., Dill, K. A. Evolution of mechanical cooperativity among myosin II motors. Proceedings of the National Academy of Sciences of the United States of America. 118 (20), 2101871118 (2021).
- Walcott, S., Warshaw, D. M., Debold, E. P. Mechanical coupling between myosin molecules causes differences between ensemble and single-molecule measurements. Biophysical Journal. 103 (3), 501-510 (2012).
- Stewart, T. J., Murthy, V., Dugan, S. P., Baker, J. E. Velocity of myosin-based actin sliding depends on attachment and detachment kinetics and reaches a maximum when myosin-binding sites on actin saturate. Journal of Biological Chemistry. 297 (5), 101178 (2021).
- Hilbert, L., Cumarasamy, S., Zitouni, N. B., Mackey, M. C., Lauzon, A. M. The kinetics of mechanically coupled myosins exhibit group size-dependent regimes. Biophysical Journal. 105 (6), 1466-1474 (2013).
- Debold, E. P., Walcott, S., Woodward, M., Turner, M. A. Direct observation of phosphate inhibiting the Force-generating capacity of a miniensemble of myosin molecules. Biophysical Journal. 105 (10), 2374-2384 (2013).
- Kaya, M., Higuchi, H. Nonlinear elasticity and an 8-nm working stroke of single myosin molecules in myofilaments. Science. 329 (5992), 686-689 (2010).
- Pertici, I., et al. A myosin II nanomachine mimicking the striated muscle. Nature Communications. 9 (1), 1-10 (2018).
- Cheng, Y. S., De Souza Leite, F., Rassier, D. E. The load dependence and the force-velocity relation in intact myosin filaments from skeletal and smooth muscles. American Journal of Physiology - Cell Physiology. 318 (1), 103-110 (2020).
- Stam, S., Alberts, J., Gardel, M. L., Munro, E. Isoforms confer characteristic force generation and mechanosensation by myosin II filaments. Biophysical Journal. 108 (8), 1997-2006 (2015).
- Rastogi, K., Puliyakodan, M. S., Pandey, V., Nath, S., Elangovan, R. Maximum limit to the number of myosin II motors participating in processive sliding of actin. Scientific Reports. 6, 1-11 (2016).
- Debold, E. P., Patlak, J. B., Warshaw, D. M. Slip sliding away: Load-dependence of velocity generated by skeletal muscle myosin molecules in the laser trap. Biophysical Journal. 89 (5), 34-36 (2005).
- Albert, P. J., Erdmann, T., Schwarz, U. S. Stochastic dynamics and mechanosensitivity of myosin II minifilaments. New Journal of Physics. 16, (2014).
- Erdmann, T., Schwarz, U. S. Stochastic force generation by small ensembles of myosin II motors. Physical Review Letters. 108 (18), 1-5 (2012).
- Guo, B., Guilford, W. H. The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motility and the Cytoskeleton. 59 (4), 264-272 (2004).
- Miller-Jaster, K. N., Petrie Aronin, C. E., Guilford, W. H. A quantitative comparison of blocking agents in the in vitro motility assay. Cellular and Molecular Bioengineering. 5 (1), 44-51 (2012).
- Mansoon, A., Balaz, M., Albet-Torres, N., Rosengren, K. J. In vitro assays of molecular motors -- impact of motor-surface interactions. Frontiers in Bioscience. 13, 5732-5754 (2008).
- Persson, M., et al. Heavy meromyosin molecules extending more than 50 nm above adsorbing electronegative surfaces. Langmuir. 26 (12), 9927-9936 (2010).
- Kron, S. J., Spudich, J. A. Fluorescent actin filaments move on myosin fixed to a glass surface. Proceedings of the National Academy of Sciences of the United States of America. 83 (17), 6272-6276 (1986).
- Yanagida, T., Nakase, M., Nishiyama, K., Oosawa, F. Direct observation of motion of single F-actin filaments in the presence of myosin. Nature. 307 (5946), 58-60 (1984).
- Tsuda, Y., Yasutake, H., Ishijima, A., Yanagida, T. Torsional rigidity of single actin filaments and actin-actin bond breaking force under torsion measured directly by in vitro micromanipulation. Proceedings of the National Academy of Sciences of the United States of America. 93 (23), 12937-12942 (1996).
- Stewart, T. J., et al. Actin sliding velocities are influenced by the driving forces of actin-myosin binding. Cellular and Molecular Bioengineering. 6 (1), 26-37 (2013).
- Harada, Y., Sakurada, K., Aoki, T., Thomas, D. D., Yanagida, T. Mechanochemical coupling in actomyosin energy transduction by in vitro movement assay. Journal of Molecular Biology. 216 (1), 49-68 (1990).
- Fordyce, P. M., Valentine, M. T., Block, S. M. Advances in surface-based assays for single molecules. Single-Molecule Techniques: A Laboratory Manual. , 431-460 (2008).
- Ozeki, T., et al. Surface-bound casein modulates the adsorption and activity of kinesin on SiO2 surfaces. Biophysical Journal. 96 (8), 3305-3318 (2009).
- Neuman, K. C., Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nature Methods. 5 (6), 491-505 (2008).
- Neuman, K. C., Block, S. M.
- Thoresen, T., Lenz, M., Gardel, M. L. Thick filament length and isoform composition determine self-organized contractile units in actomyosin bundles. Biophysical Journal. 104 (3), 655-665 (2013).
- Matusovsky, O. S., et al. Millisecond conformational dynamics of skeletal Myosin II power stroke studied by high-speed atomic force microscopy. ACS Nano. 15 (2), 2229-2239 (2021).
- Reinemann, D. N., et al. Collective force regulation in anti-parallel microtubule gliding by dimeric Kif15 kinesin motors. Current Biology. 27 (18), 2810-2820 (2017).
- Cordova, J. C., et al. Bioconjugated core-shell microparticles for high-force optical trapping. Particle and Particle Systems Characterization. 35 (3), 1-8 (2018).
- Reinemann, D. N., Norris, S. R., Ohi, R., Lang, M. J. Processive Kinesin-14 HSET exhibits directional flexibility depending on motor traffic. Current Biology. 28 (14), 2356-2362 (2018).
- Forth, S., Hsia, K. C., Shimamoto, Y., Kapoor, T. M. Asymmetric friction of nonmotor MAPs can lead to their directional motion in active microtubule networks. Cell. 157 (2), 420-432 (2014).
- Shimamoto, Y., Kapoor, T. M. Analyzing the micromechanics of the cell division apparatus. Methods in Cell Biology. 145, 173-190 (2018).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Thoresen, T., Lenz, M., Gardel, M. L.
- Murrell, M., Thoresen, T., Gardel, M.
- Weirich, K. L., Stam, S., Munro, E., Gardel, M. L. Actin bundle architecture and mechanics regulate myosin II force generation. Biophysical Journal. 120 (10), 1957-1970 (2021).
