면역침전반응 기반 기술: 아가로스 비즈를 사용한 내인성 단백질의 정제
1. 단백질 A/G 플러스 아가로즈 구슬을 이용한 면역 침전
세포 리세이트 제제
- 심심분리기 108 티모세포는 마이크로센심분리기에서 3분 동안 13,000rpm에 상복부를 제거합니다.
참고: 세포 수는 원하는 단백질과 선택한 세포 유형의 발현 수준에 따라 달라집니다. - PMSF를 통해 500 μL 용해 버퍼 RIPA에서 세포를 다시 일시 중단합니다.
- 소용돌이로 몇 가지 빠른 펄스를 사용하여 세포를 방해한 다음 주사기에 부착 된 25 G 바늘로 lysate을 몇 번 흡입하십시오.
참고: 거품을 만들지 마십시오. 더 큰 세포 모형을 위한 21G 바늘과 같은 더 큰 바늘을 사용하십시오. - 10 분 동안 얼음에 세포 용해를 배양.
- 4°C에서 15분 동안 13,000rpm의 lysate원심분리기.
- 상체를 신선한 마이크로 센심 분리기 튜브로 옮기십시오.
사전 클리어링
- 20 μL 단백질 A/G PLUS 아가로즈 구슬과 등류형 대조군 항체의 1 μg(여기, 마우스 IgG1 동형 대조군 항체가 사용됨)를 용액에 첨가한다.
참고: 사용되는 동형 항체의 선택은 풀다운 단계에서 나중에 사용되는 단백질 특이적 항체에 의존할 것이다. - 차가운 방(4°C)의 원심분리기 회전기에 lysate 믹스를 30분 동안 배양합니다.
- 4°C에서 30s의 3200 rpm에서 샘플을 원심분리합니다.
- 미리 클리어된 상체를 1.5mL 마이크로센심분리기 튜브로 옮기습니다. 펠릿을 버리십시오.
단백질 농도 측정
- 브래드포드 분석능력을 수행하여 세포 의 단백질 농도를 결정한다.
- Aliquot 1000 μL 브래드포드 시약7 마이크로 센심 분리기 튜브로.
- 다음과 같은 양의 BSA 단백질 표준(2 mg/mL)을 튜브 6개(표 1)에 넣습니다.
| 튜브 번호 | BSA 부피(μL) (2 mg/mL) | 단백질 농도(μg/μL) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
표 1: BSA 단백질 표준 금액
- 7번째 튜브에서 미리 지워진 lysate의 1 μL을 추가합니다.
참고: 샘플 농도가 분석 감지 범위 내에 있는지 확인하려면 1:2 또는 1:5 용액 희석을 준비하고 분석합니다. - 7개의 튜브 각각에서 200 μL을 평평한 바닥의 개별 우물에 넣고, 96웰 플레이트는 각 샘플을 삼중으로 반복합니다.
- 595 nm에서 플레이트 리더의 접시를 읽으십시오.
- 표준 곡선을 엑셀로 생성하고 미리 지워진 lysate의 단백질 농도를 계산합니다.
아래로 당겨
- 이 예에서 c-myc인 '제어'로 두 개의 신선한 1.5 mL 마이크로 원심 분리튜브를 '제어'로 표시하고 다른 하나는 '테스트'로 레이블을 지정합니다.
- 미리 지워진 용해 500μg를 각 튜브에 넣습니다.
참고: 여기서 사용되는 단백질의 양은 정제되기를 원하는 단백질의 양에 따라 달라집니다. - 리시스 버퍼를 사용하여 각 튜브의 총 부피를 최대 500 μL까지 끌어올수 있습니다.
- 시험단관에 항-c-myc 항체 2μg을 추가하고 2μg 마우스 IgG1 이소타입 대조군 항체를 대조군에 첨가한다.
참고: 항체의 양은 항체 효능 및 표적 단백질의 양에 따라 달라집니다. - 차가운 방 (4 °C)의 회전에 튜브를 2 시간 동안 배양하십시오.
- 각 튜브에 20 μL 단백질 A/G PLUS 아가로즈 구슬을 추가합니다.
참고: 구슬의 손상을 방지하기 위해 끝 차단된 파이펫 팁을 사용하는 것이 좋습니다. - 하룻밤 동안 차가운 방 (4 °C)에서 회전에 배양하십시오.
참고: 표적 단백질및 항체 효능에 따라, 이 단계는 1시간에서 하룻밤사이에 달라질 수 있다. - 30s 4°C의 경우 3200rpm의 튜브를 원심분리하여 구슬을 끌어당깁니다.
- 각 튜브에서 슈퍼네티드를 흡인합니다.
참고: 표적 단백질은 이제 구슬에 묶여 있습니다. - 500 μL 1X 덜벡코의 PBS를 사용하여 구슬을 두 번 세척합니다.
- 30 s 4 °C의 경우 3200 rpm의 튜브원심분리기.
참고: 보다 엄격한 세척을 위해 RIPA와 같은 보다 엄격한 버퍼를 사용하십시오. - 각 튜브에서 버퍼를 흡인합니다. 젤 로딩 팁을 사용하여 구슬에서 남은 버퍼를 제거하고 구슬을 얼음위에 두어 단백질을 엘로우시합니다.
참고: 이 예에서, 단백질은 서양 얼룩 분석을 위해 구슬을 끓여서 SDS-PAGE 실행 버퍼로 용출됩니다. 이 접근법은 IP 결과를 확인하거나 단백질 단백질 상호 작용을 검사하는 데 적합합니다. 구조적 또는 효소 분석을 위한 단백질의 정제와 같은 다른 다운스트림 응용 분야의 경우, 에피토프 태그(플래그 태그 또는 myc-tag)와 같은 보다 정교한 시스템은 관심 있는 단백질을 가진 항체의 용출을 피하기 위해 사용된다.
2. 웨스턴 블롯 분석을 통한 IP 검증
SDS 페이지 전기 포레시스:
- 20 μL SDS-PAGE 로딩 염료에서 β-메카토-에토놀을 포함하는 비드를 다시 일시 중단합니다.
- 샘플을 95°C에서 5분간 끓입니다.
- 원심 분리구는 실온에서 10 초 동안 13,000 rpm에서 구슬을 원심분리합니다.
- 젤 로딩 팁을 사용하여 구슬에서 얻은 샘플을 조심스럽게 파이프화하여 4-15 % 그라데이션 SDS-PAGE 젤의 우물에 로드하십시오.
- 샘플 외에도 단백질 사다리가 있는 차선과 미리 지워진 리세이트가 있는 차선을 적재하여 로딩 컨트롤로 작용합니다.
- 염료 전면이 젤의 바닥에 도달 할 때까지 100 V에서 실행 (~1h).
웨스턴 블롯 분석:
- PVDF 멤브레인이 젤과 붉은 음극 사이에 있는지 확인, 웨스턴 블롯 샌드위치를 확인합니다.
- 100 V에서 1 시간 동안 전송합니다.
- 낮은 설정에서 로커에 1 h에 대한 실온에서 5 mL 차단 버퍼에 멤브레인을 배치, 비 특정 단백질 결합 부위를 차단합니다.
참고: 블로킹 버퍼, 1차 항체, 이차 항체 및 세차의 부피가 더 큰 크기의 블롯에 대해 증가해야 할 수도 있다. - 블로우를 5mL 항-c-myc 항체로 낮은 설정에서 로커의 4°C에서 밤새 블로킹 버퍼에 배양합니다.
참고: 여기에 사용되는 항체는 풀다운 단계에서 사용되는 항체와 다릅니다. - 5mL TBST를 사용하여 블로트 3-6번 세척할 수 있으며, 각 세척은 낮은 설정에서 로커의 실온에서 5분입니다.
- HRP 태그된 래키 안티래빗 라이트 체인 이차 항체로 블롯을 버퍼블로 배양하고, 낮은 환경에서 로커의 실온에서 1h를 배양한다.
참고: 이차 항체의 선택은 서양 얼룩에 사용되는 1 차적인 항체에 달려 있습니다. 또한, 표적 단백질이 항체의 중사슬에 분자량이 가깝기 때문에 특정 이차광사슬이 프로토콜에 사용된다. 표적 단백질이 50kDa에 가까우면, 라이트 체인 특정 이차를 사용하십시오. 표적 단백질이 25kDa에 가까운 경우, 사용 및 중고 사슬 특정 이차. - 5mL TBST를 사용하여 블로트 3-6번 세척할 수 있으며, 각 세척은 낮은 설정에서 로커의 실온에서 5분입니다.
- 여분의 액체를 제거하기 위해 실험실 와이프에 얼룩에서 액체를 제거하고 얼룩의 가장자리를 두드려.
- 1x 화학 발광 검출 시약으로 얼룩을 덮고 1 분 동안 배양하십시오.
참고: 감지 시약이 가볍고 시간에 민감하기 때문에 다음 단계를 신속하게 수행해야 합니다. - 실험실 와이프에 블롯의 가장자리를 두드려 과도한 검출 시약을 제거합니다.
- 이미징 표면에 블롯을 놓습니다.
참고: 케미발광 얼룩도 필름을 사용하여 시각화할 수 있습니다. - 'Chemiluminescent 프로그램'을사용하여 이미지를 사용하여 10초에서 5분까지 여러 시간 지점을 캡처합니다.
참고: 최적의 시간은 단백질 양과 항체의 품질에 따라 변경될 수 있습니다. - 최적의 밴드 가시성을 가진 이미지를 선택한 다음 해당 이미지를 내보냅니다.
- 블롯을 이동하기 전에 이미저를 사용하여 블로트 사진을 찍어 사다리의 위치를 캡처합니다. 그런 다음 해당 이미지도 내보냅니다.
- 슬라이드 준비 소프트웨어(예: 파워포인트)를 사용하여 밴드와 래더 이미지를 정렬하여 단일 이미지를 형성합니다.
출처: 수잔나 C. 시슬러1,토냐 J. 웹1
1 미생물학 및 면역학학과, 메릴랜드 대학교 볼티모어, MD 21201
면역 침전물(IP, '풀다운' 분석이라고도 함)은 다양한 분야에서 응용 분야를 가지고 있는 널리 사용되는 기술이다. 1984년에 처음 구상된 이 요리는 1988년(1, 2)에 정제되었다. IP의 근본적인 목표는 그 단백질에 대하여 항체를 사용하여 특정 단백질의 정제 그리고 격리입니다. 단어 "면역"은 "강수량"이라는 단어가 용액에서 특정 물질을 당기는 것을 의미하는 동안 항체의 사용을 지칭한다. 표적 단백질은 내인성 또는 재조합일 지도 모릅니다. 대부분의 재조합 단백질에는 후속 정화를 단순화하기 위해 에피토프 태그(즉, myc 또는 flag)가 부착되어 있습니다. 전형적으로, 재조합 에피토프 태그에 대한 항체가 매우 강하고 효과적이기 때문에 재조합 단백질 IP를 최적화하는 것이 더 쉽습니다. 내인 성 단백질에 대한 항체는 매우 가변적 인 효능을 가지고 있어 이러한 IP를 최적화하기가 훨씬 더 어려워지게됩니다. 면역 침전 후 필요한 단계는 정화의 검증이다. 격리된 단백질은 SDS-PAGE를 사용하여 해결되고 그 후 서양 얼룩에 의한 순도를 위해 조사됩니다(그림 1). 중요한 대조군은 정확한 단백질의 당김을 확인하기 위해 서양 블롯 동안 다른 항체를 사용하는 것이다. IP와 후속 기술의 조합은 강력한 분석 도구입니다. 정제 후의 목표는 NMR, 질량 분광법 및 시험관 내 분석, 또는 단백질의 상호 작용 파트너(즉, 단백질, DNA, RNA)의 분석(즉, 단백질, DNA, RNA) (3, 4, 5)에 의해 단백질 자체의 특성화일 수 있다.
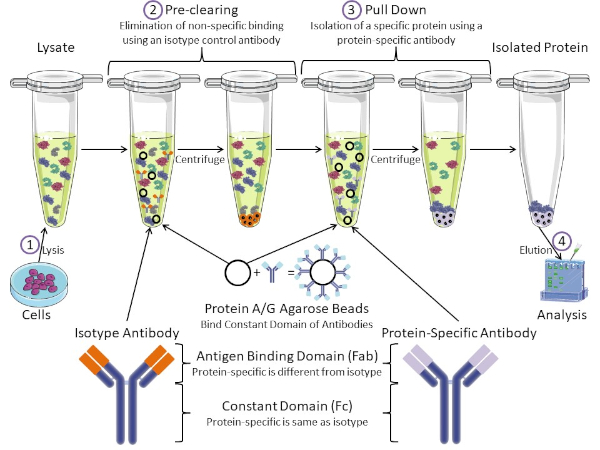
그림 1: 면역 침전 절차의 개요. 면역 침전은 항체를 사용하여 특정 단백질의 격리이다. 세포로부터 용재를 생산한 후, 두 가지 주요 단계-사전 정리 및 풀다운이 있습니다. 사전 클리어링 단계 동안, 세포-lysates는 isotype 대조군 항체를 사용하여 비특이적으로 항체에 결합하는 단백질의 미리 지워집니다. 당기면, 표적 단백질은 단백질 특이적 항체를 사용하여 아래로 당겨졌다. 고립 된 단백질은 서양 얼룩에 의해 분석됩니다. 동형 항체 및 단백질 특이적 항체는 동일한 상수 도메인을 가지지만, 상이한 항원 결합 도메인이 있다. 이 프로토콜의 핵심 성분은 항체의 일정한 도메인을 결합하는 단백질 A/G 아가로즈 구슬입니다 - 표적 단백질의 면역 침전을 허용합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
항체는 다른 형태의 단백질 정제(즉, 니켈 친화 컬럼 정제)와 구별되는 면역 침전의 핵심 성분이다. 항체는 특정 단백질 에피토프를 인식할 수 있는 B 세포에 의해 만들어진 분자입니다. 항체에는 상수(Fc) 및 항원 결합(Fab)(도 1)의 두 가지 영역이 있습니다. 상수 도메인은 항체의 유형을 식별하고 생체 내에서기능을 지시한다. 일반적으로 IP에 사용되는 항체의 상수 영역은 마우스, 쥐 또는 토끼 IgG입니다. 항체의 항원 결합 부는 특정 단백질의 특정 에피토프를 인식한다. 항체는 단백질이 변성될 때 존재하지 않을 수 있는 접힌 단백질에 대한 전형체를 인식할 수 있고 그 반대의 경우도 마찬가지입니다. 따라서, 에피토프의 가용성은 단백질 접이식에 달려 있습니다 - IP에 대한 항체 및 조건을 선택할 때 고려해야 할 중요한 요소를 식별합니다.
대핵계와 진핵계에는 항체 결합 단백질이 있습니다. 진핵 계통에서, 목적은 근생 계통에 있는 동안 박테리아로부터 면역 보호입니다, 목적은 면역 계통으로부터 보호입니다. 항체 결합 단백질은 두 가지 방법으로 IP 방법론에 영향을 미칩니다. 첫째, 항체를 결합하는 단백질의 용액을 제거하는 데 필요한 사전 정리 단계(도 1)가 있어 최종 제품에 비특이적 결합을 줄입니다. 이 단계는 단백질 특이적 항체와 는 다른 항체 결합 도메인과 동일한 상수 영역을 가지고 있는 동종형 항체를 사용합니다. 세균성 항체 결합 단백질은 이 방법의 두 번째 핵심 성분이다. 단백질 특이적 항체가 표적 단백질을 결합한 후, 항체: 단백질 복합체는 아래로 당겨져야 한다(도 1). 단백질 A, G 및 L은 항체의 일정한 도메인을 결합하는 세균성 단백질입니다. 박테리아는 면역 계통을 전복하기 위하여 이것을 사용하는 동안, 연구원은 쉬운 항체 정화를 위한 이 시스템을 공동 선택했습니다, 그리고 사전 정리 및 풀다운 단계 둘 다 도중 이용됩니다. 이 단백질은 다른 종 및 다른 일정한 도메인 하위형에 대한 상이한 결합 친화성을 가지고 있습니다 - IP를 위한 조건을 선택할 때 고려해야 할 또 다른 요인. 많은 회사에서 단백질 A/G 라벨아가로즈 구슬(그림 1), 미리 만들어진 스핀 컬럼 또는 수지를 판매하여 컬럼을 만듭니다. 일반적으로 구슬과 스핀 컬럼은 더 작은 샘플 크기에 사용되는 반면 수지는 대량 정화에 사용됩니다.
이 실험실 운동에서, 우리는 단백질 A/G 플러스 아가로즈 구슬 기지를 둔 기본적인 면역 침전 기술을 사용하여, 1 차적인 뮤린 흉구에서 내인성 단백질 c-myc를 정화하는 방법을 보여줍니다. 프로토콜은 세포 용해 준비에서 시작하고 서양 얼룩 분석을 사용하여 성공적인 단백질 풀다운의 검증으로 끝납니다.
1. 단백질 A/G 플러스 아가로즈 구슬을 이용한 면역 침전
세포 리세이트 제제
- 심심분리기 108 티모세포는 마이크로센심분리기에서 3분 동안 13,000rpm에 상복부를 제거합니다.
참고: 세포 수는 원하는 단백질과 선택한 세포 유형의 발현 수준에 따라 달라집니다. - PMSF를 통해 500 μL 용해 버퍼 RIPA에서 세포를 다시 일시 중단합니다.
- 소용돌이로 몇 가지 빠른 펄스를 사용하여 세포를 방해한 다음 주사기에 부착 된 25 G 바늘로 lysate을 몇 번 흡입하십시오.
참고: 거품을 만들지 마십시오. 더 큰 세포 모형을 위한 21G 바늘과 같은 더 큰 바늘을 사용하십시오. - 10 분 동안 얼음에 세포 용해를 배양.
- 4°C에서 15분 동안 13,000rpm의 lysate원심분리기.
- 상체를 신선한 마이크로 센심 분리기 튜브로 옮기십시오.
사전 클리어링
- 20 μL 단백질 A/G PLUS 아가로즈 구슬과 등류형 대조군 항체의 1 μg(여기, 마우스 IgG1 동형 대조군 항체가 사용됨)를 용액에 첨가한다.
참고: 사용되는 동형 항체의 선택은 풀다운 단계에서 나중에 사용되는 단백질 특이적 항체에 의존할 것이다. - 차가운 방(4°C)의 원심분리기 회전기에 lysate 믹스를 30분 동안 배양합니다.
- 4°C에서 30s의 3200 rpm에서 샘플을 원심분리합니다.
- 미리 클리어된 상체를 1.5mL 마이크로센심분리기 튜브로 옮기습니다. 펠릿을 버리십시오.
단백질 농도 측정
- 브래드포드 분석능력을 수행하여 세포 의 단백질 농도를 결정한다.
- Aliquot 1000 μL 브래드포드 시약7 마이크로 센심 분리기 튜브로.
- 다음과 같은 양의 BSA 단백질 표준(2 mg/mL)을 튜브 6개(표 1)에 넣습니다.
| 튜브 번호 | BSA 부피(μL) (2 mg/mL) | 단백질 농도(μg/μL) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
표 1: BSA 단백질 표준 금액
- 7번째 튜브에서 미리 지워진 lysate의 1 μL을 추가합니다.
참고: 샘플 농도가 분석 감지 범위 내에 있는지 확인하려면 1:2 또는 1:5 용액 희석을 준비하고 분석합니다. - 7개의 튜브 각각에서 200 μL을 평평한 바닥의 개별 우물에 넣고, 96웰 플레이트는 각 샘플을 삼중으로 반복합니다.
- 595 nm에서 플레이트 리더의 접시를 읽으십시오.
- 표준 곡선을 엑셀로 생성하고 미리 지워진 lysate의 단백질 농도를 계산합니다.
아래로 당겨
- 이 예에서 c-myc인 '제어'로 두 개의 신선한 1.5 mL 마이크로 원심 분리튜브를 '제어'로 표시하고 다른 하나는 '테스트'로 레이블을 지정합니다.
- 미리 지워진 용해 500μg를 각 튜브에 넣습니다.
참고: 여기서 사용되는 단백질의 양은 정제되기를 원하는 단백질의 양에 따라 달라집니다. - 리시스 버퍼를 사용하여 각 튜브의 총 부피를 최대 500 μL까지 끌어올수 있습니다.
- 시험단관에 항-c-myc 항체 2μg을 추가하고 2μg 마우스 IgG1 이소타입 대조군 항체를 대조군에 첨가한다.
참고: 항체의 양은 항체 효능 및 표적 단백질의 양에 따라 달라집니다. - 차가운 방 (4 °C)의 회전에 튜브를 2 시간 동안 배양하십시오.
- 각 튜브에 20 μL 단백질 A/G PLUS 아가로즈 구슬을 추가합니다.
참고: 구슬의 손상을 방지하기 위해 끝 차단된 파이펫 팁을 사용하는 것이 좋습니다. - 하룻밤 동안 차가운 방 (4 °C)에서 회전에 배양하십시오.
참고: 표적 단백질및 항체 효능에 따라, 이 단계는 1시간에서 하룻밤사이에 달라질 수 있다. - 30s 4°C의 경우 3200rpm의 튜브를 원심분리하여 구슬을 끌어당깁니다.
- 각 튜브에서 슈퍼네티드를 흡인합니다.
참고: 표적 단백질은 이제 구슬에 묶여 있습니다. - 500 μL 1X 덜벡코의 PBS를 사용하여 구슬을 두 번 세척합니다.
- 30 s 4 °C의 경우 3200 rpm의 튜브원심분리기.
참고: 보다 엄격한 세척을 위해 RIPA와 같은 보다 엄격한 버퍼를 사용하십시오. - 각 튜브에서 버퍼를 흡인합니다. 젤 로딩 팁을 사용하여 구슬에서 남은 버퍼를 제거하고 구슬을 얼음위에 두어 단백질을 엘로우시합니다.
참고: 이 예에서, 단백질은 서양 얼룩 분석을 위해 구슬을 끓여서 SDS-PAGE 실행 버퍼로 용출됩니다. 이 접근법은 IP 결과를 확인하거나 단백질 단백질 상호 작용을 검사하는 데 적합합니다. 구조적 또는 효소 분석을 위한 단백질의 정제와 같은 다른 다운스트림 응용 분야의 경우, 에피토프 태그(플래그 태그 또는 myc-tag)와 같은 보다 정교한 시스템은 관심 있는 단백질을 가진 항체의 용출을 피하기 위해 사용된다.
2. 웨스턴 블롯 분석을 통한 IP 검증
SDS 페이지 전기 포레시스:
- 20 μL SDS-PAGE 로딩 염료에서 β-메카토-에토놀을 포함하는 비드를 다시 일시 중단합니다.
- 샘플을 95°C에서 5분간 끓입니다.
- 원심 분리구는 실온에서 10 초 동안 13,000 rpm에서 구슬을 원심분리합니다.
- 젤 로딩 팁을 사용하여 구슬에서 얻은 샘플을 조심스럽게 파이프화하여 4-15 % 그라데이션 SDS-PAGE 젤의 우물에 로드하십시오.
- 샘플 외에도 단백질 사다리가 있는 차선과 미리 지워진 리세이트가 있는 차선을 적재하여 로딩 컨트롤로 작용합니다.
- 염료 전면이 젤의 바닥에 도달 할 때까지 100 V에서 실행 (~1h).
웨스턴 블롯 분석:
- PVDF 멤브레인이 젤과 붉은 음극 사이에 있는지 확인, 웨스턴 블롯 샌드위치를 확인합니다.
- 100 V에서 1 시간 동안 전송합니다.
- 낮은 설정에서 로커에 1 h에 대한 실온에서 5 mL 차단 버퍼에 멤브레인을 배치, 비 특정 단백질 결합 부위를 차단합니다.
참고: 블로킹 버퍼, 1차 항체, 이차 항체 및 세차의 부피가 더 큰 크기의 블롯에 대해 증가해야 할 수도 있다. - 블로우를 5mL 항-c-myc 항체로 낮은 설정에서 로커의 4°C에서 밤새 블로킹 버퍼에 배양합니다.
참고: 여기에 사용되는 항체는 풀다운 단계에서 사용되는 항체와 다릅니다. - 5mL TBST를 사용하여 블로트 3-6번 세척할 수 있으며, 각 세척은 낮은 설정에서 로커의 실온에서 5분입니다.
- HRP 태그된 래키 안티래빗 라이트 체인 이차 항체로 블롯을 버퍼블로 배양하고, 낮은 환경에서 로커의 실온에서 1h를 배양한다.
참고: 이차 항체의 선택은 서양 얼룩에 사용되는 1 차적인 항체에 달려 있습니다. 또한, 표적 단백질이 항체의 중사슬에 분자량이 가깝기 때문에 특정 이차광사슬이 프로토콜에 사용된다. 표적 단백질이 50kDa에 가까우면, 라이트 체인 특정 이차를 사용하십시오. 표적 단백질이 25kDa에 가까운 경우, 사용 및 중고 사슬 특정 이차. - 5mL TBST를 사용하여 블로트 3-6번 세척할 수 있으며, 각 세척은 낮은 설정에서 로커의 실온에서 5분입니다.
- 여분의 액체를 제거하기 위해 실험실 와이프에 얼룩에서 액체를 제거하고 얼룩의 가장자리를 두드려.
- 1x 화학 발광 검출 시약으로 얼룩을 덮고 1 분 동안 배양하십시오.
참고: 감지 시약이 가볍고 시간에 민감하기 때문에 다음 단계를 신속하게 수행해야 합니다. - 실험실 와이프에 블롯의 가장자리를 두드려 과도한 검출 시약을 제거합니다.
- 이미징 표면에 블롯을 놓습니다.
참고: 케미발광 얼룩도 필름을 사용하여 시각화할 수 있습니다. - 'Chemiluminescent 프로그램'을사용하여 이미지를 사용하여 10초에서 5분까지 여러 시간 지점을 캡처합니다.
참고: 최적의 시간은 단백질 양과 항체의 품질에 따라 변경될 수 있습니다. - 최적의 밴드 가시성을 가진 이미지를 선택한 다음 해당 이미지를 내보냅니다.
- 블롯을 이동하기 전에 이미저를 사용하여 블로트 사진을 찍어 사다리의 위치를 캡처합니다. 그런 다음 해당 이미지도 내보냅니다.
- 슬라이드 준비 소프트웨어(예: 파워포인트)를 사용하여 밴드와 래더 이미지를 정렬하여 단일 이미지를 형성합니다.
면역침전(Immunoprecipitation, IP)은 단백질 특성 분석을 위해 세포나 조직 용해물 또는 체액에서 관심 단백질을 분리하거나 단백질과 단백질의 상호작용을 조사하기 위해 널리 사용되는 기법입니다.
이 과정은 표적 단백질에 대한 친화력과 특이성이 높은 항체에서 시작됩니다. 이 항체는 샘플과 혼합되어 항체-표적 복합체가 형성될 수 있습니다. 표적 단백질에 결합된 모든 단백질은 이 과정에서 항체에 간접적으로 부착됩니다. 다음으로, 용액을 박테리아 단백질에 접합 된 아가로 로즈 비드로 배양하는데, 이는 항체의 일정한 영역에 대한 강한 친화력을 갖는다. 박테리아 단백질은 항체에 결합하고 항체 표적 복합체를 비드에 연결합니다. 그런 다음, 용액을 원심분리하여 비드를 침전시켜 결합 항체, 표적 단백질 및 상호 작용하는 단백질을 포함하는 전체 복합체를 추출합니다. 마지막으로, 결합된 단백질은 비드에서 추출되어 서로 분리되어 웨스턴 블로팅(Western blotting)과 같은 기술에 의한 추가 분석에 사용됩니다.
이 기술의 여러 부분의 여러 변형이 일반적으로 사용되며, 사전 투명화, 펩타이드 태그 또는 마그네틱 비드 사용 또는 기타 비단백질 결합 파트너 분석과 같이 사용됩니다. IP는 샘플에서 비특이적 항체 결합 단백질을 제거하고 배경을 최소화하기 위해 사전 투명화 단계를 선행할 수 있습니다. 여기에는 먼저 동형 대조 항체로 샘플을 배양하여 이러한 단백질에 결합할 수 있도록 한 다음 아가로스 비드를 사용하여 복합체를 침전시키는 작업이 포함됩니다. 그러면 샘플이 실제 IP로 진행할 준비가 됩니다.
펩타이드 태그는 IP에 대해 특정 항체를 사용할 수 없는 경우에 유용합니다. 여기서, 표적 단백질은 펩타이드 에피토프 태그를 포함하도록 유전적으로 변형될 수 있으며, 태그에 대한 항체는 관심 단백질을 추출할 수 있습니다. 표적을 침전시키기 위해 아가로스 대신 마그네틱 비드가 자주 사용됩니다. 항체 표적 복합체에 결합한 후 샘플 튜브를 강한 자기장에 배치하여 용액에서 비드를 추출합니다. 따라서 원심분리의 필요성이 제거되고 속도와 편의성이 향상됩니다.
면역침전은 DNA 또는 RNA 결합 단백질을 연구하는 데에도 사용되며 각각 염색질 면역침전(chromatin immunoprecipitation) 및 RNA 면역침전(RNA immunoprecipitation)으로 알려져 있습니다. 이러한 변형은 문제를 해결하고 다양한 실험 응용 분야에 맞게 방법을 조정하는 데 유용합니다. 이 동영상에서는 세포 용해물을 사전 투명화하고 면역침전을 수행하여 관심 단백질을 추출한 후 웨스턴 블롯 분석을 수행하여 실험을 검증하는 방법을 관찰합니다.
먼저 사전 수집된 세포를 마이크로 원심분리기에 넣고 13,000rpm으로 3분 동안 회전합니다. 스핀 후 상층액을 제거한 다음 PMSF를 사용하여 500마이크로리터의 용해 완충액 RIPA에 세포를 재현탁합니다. 이제 소용돌이로 몇 번의 빠른 펄스를 사용하여 세포를 파괴한 다음 주사기에 부착된 25게이지 바늘로 용해물을 몇 번 흡인하고 거품이 생성되지 않도록 주의하십시오. 세포를 얼음 위에 15분 동안 놓습니다. 얼음 위에서 샘플을 배양한 후 용해물을 섭씨 4도에서 15분 동안 원심분리합니다.
새 1.5ml 마이크로 원심분리기 튜브에 라벨을 붙입니다. 스핀 후 상등액을 새로 라벨이 붙은 튜브로 옮기고 펠릿을 버립니다. 다음으로, 20마이크로리터의 Protein A/G PLUS-아가로스 비드와 1마이크로그램의 동형대조군 항체를 용해물(이 예에서는 마우스 IgG1 동형)에 추가하여 아가로스 비드 또는 1차 항체에 비특이적으로 결합하는 오염물질의 용해물을 미리 세척합니다. 차가운 방의 회전자에 튜브를 30분 동안 배양합니다. 냉장실에서 용해물을 30분 동안 회전시킨 후 섭씨 4도에서 30초 동안 3200rpm으로 샘플을 원심분리합니다. 원심분리기에서 튜브를 제거하고 미리 세척된 상등액을 라벨이 붙은 새 1.5ml 마이크로 원심분리기 튜브로 옮깁니다. 펠릿을 버리십시오.
이제 Bradford 분석을 수행하여 세포 용해물의 단백질 농도를 측정합니다. 라벨 7 1. 5밀리리터 마이크로 원심분리기 튜브 1에서 6까지 그리고 1000마이크로리터의 Bradford 시약을 각 튜브에 샘플링 및 분취합니다. 6개의 튜브는 각 튜브에 알려진 다양한 양의 BSA를 추가하여 표준 곡선을 만드는 데 사용됩니다. 추가할 금액이 이 표에 나열되어 있습니다. 일곱 번째 샘플 튜브에 미리 세척된 용해물 1마이크로리터를 추가합니다. 7개의 튜브 각각에서 200마이크로리터를 평평한 바닥 96웰 플레이트의 개별 웰에 넣고 각 샘플을 세 번 반복하여 7개의 샘플로 구성된 3개의 열이 있도록 합니다. 595나노미터의 파장을 사용하여 플레이트 리더에서 플레이트를 읽습니다. Excel에서 표준 곡선을 만든 후 미리 세척된 용해물의 단백질 농도를 계산합니다.
다음으로, 두 개의 1.5ml 마이크로 원심분리기 튜브(하나는 대조군으로, 다른 하나는 테스트용)로 표시하며, 이 예에서는 c-myc 항체가 됩니다. 사전 세척된 용해물 500마이크로그램을 각 튜브에 넣은 다음 용해 완충액을 사용하여 각 튜브의 총 부피를 최대 500마이크로리터까지 가져옵니다. 다음으로, 항-c-myc 항체 2마이크로그램을 검사군 튜브에 추가합니다. 대조군의 경우 마우스 IgG1 isotype 대조군 항체 2마이크로그램을 추가합니다. 항체가 튜브에 추가되면 샘플을 차가운 방의 회전기에 놓고 2시간 동안 배양합니다. 이제 아가로스 구슬을 추가합니다. 이렇게 하려면 피펫 팁의 끝을 잘라낸 다음 이 수정된 팁을 사용하여 각 튜브에 200마이크로리터의 Protein A/G PLUS-아가로스 비드를 추가하는 것이 좋습니다. 밤새 차가운 방의 회전식에서 튜브를 배양합니다.
배양 후 회전기에서 튜브를 제거하고 마이크로 원심분리기에서 용해물을 회전시켜 비드를 아래로 당깁니다. 스핀이 완료되면 원심분리기에서 튜브를 제거하고 각 튜브에서 상층액을 흡인합니다. 다음으로, 500마이크로리터의 1X Dulbecco의 PBS를 사용하여 비드를 세척합니다. 튜브를 마이크로 원심분리기에 넣고 섭씨 4도에서 30초 동안 회전합니다. 그런 다음 상층액을 제거하십시오. 세척 및 원심분리 단계를 한 번 더 반복하여 총 두 번씩 반복합니다. 마이크로 원심분리기에서 튜브를 제거하고 각 튜브에서 버퍼를 흡입합니다. 겔 로딩 팁을 사용하여 비드에 남아 있는 완충액을 제거하고 비드를 얼음 위에 유지하여 결합된 단백질을 용리시킵니다.
이 예에서 단백질은 웨스턴 블롯 분석을 위해 끓여서 SDS-PAGE 러닝 버퍼로 용리됩니다. 이를 위해 베타-메르캅토에탄올 또는 BME를 함유한 20마이크로리터의 SDS-PAGE 로딩 염료에 비드를 재현탁합니다. 샘플을 섭씨 95도에서 5분 동안 끓여서 비드에서 면역복합체를 분리합니다. 그런 다음 실온에서 10초 동안 최대 속도로 비드를 원심분리합니다. 마이크로 원심분리기에서 튜브를 제거하고 실온의 랙에 보관합니다. 겔 로딩 팁을 사용하여 비드의 샘플을 조심스럽게 피펫팅하고 4-15% 구배 SDS-PAGE 겔의 웰에 로드합니다. 샘플 외에도, 단백질 ladder가 있는 레인과 사전 세척된 용해물이 있는 레인을 로딩하여 로딩 제어 역할을 합니다. 젤이 로드되면 젤을 100볼트에서 실행합니다.
염료 선단이 겔 바닥에 도달한 후(약 1시간 소요) 겔을 멈추고 웨스턴 블롯 샌드위치를 만들어 PVDF 멤브레인이 겔과 음극 사이에 있는지 확인합니다. 웨스턴 블롯 샌드위치를 전사 장치에 넣고 겔의 단백질을 100볼트에서 1시간 동안 멤브레인으로 옮깁니다. 전달이 완료된 후 항체가 멤브레인에 비특이적으로 결합하는 것을 방지하기 위해 멤브레인을 5ml 블록에 놓습니다. 실온에서 한 시간 동안 낮은 설정으로 흔들어주세요. 타이머가 울리면 차단 버퍼를 제거합니다. 검출 항체가 있는 차단 완충액 5ml를 멤브레인에 추가합니다. 여기서, 풀다운에 사용된 것과 다른 항-c-myc 항체가 사용된다.
밤새 섭씨 4도의 낮은 설정의 로커에서 블롯을 배양합니다. 배양 후 항체와 차단 완충액을 제거합니다. TBST 5ml를 사용하여 실온에서 5분 동안 낮은 설정의 로커에서 얼룩을 씻습니다. 이 세척 단계는 총 3-6회 세척에 대해 2-5회 반복해야 하며, 각 세척에 대해 새로운 TBST를 사용해야 합니다. 5ml의 1-1000 2차 항체와 차단 완충액을 블롯에 추가합니다. 이 경우, 2차 항체는 HRP 태그가 붙은 안티 래빗 경쇄입니다. 실온에서 하나를 위해 낮은 설정에서 로커의 블롯을 배양합니다. 다음으로, 버퍼를 제거하고 TBST 5ml로 블롯을 세척합니다. 이 세척물을 실온에서 5분 동안 낮은 설정으로 로커에 배양하십시오. 이 세척을 총 6-12회 반복하며 각 세척에는 5ml의 TBST를 새로 넣습니다. 먼저 얼룩에서 액체를 부어 최종 세척을 제거합니다. 그런 다음 핀셋을 사용하여 실험실 물티슈에 얼룩 가장자리를 두드려 여분의 액체를 제거한 다음 얼룩을 새 용기에 넣습니다. 다음으로, 1X 화학발광 검출 시약으로 블롯을 덮고 1분 동안 배양합니다.
빠르게 작업하여 실험실 청소포의 블롯 가장자리를 두드려 과도한 검출 시약을 제거한 다음 Imager 트레이의 이미징 표면에 블롯을 놓습니다. 화학발광 프로그램을 사용하여 10초에서 30초까지 여러 시간 지점을 캡처한 이미지. 블롯을 이미지화한 후 최적의 밴드 가시성을 가진 이미지를 선택한 다음 해당 이미지를 내보냅니다. 블롯을 이동하기 전에 이미저를 사용하여 블롯 사진을 찍어 사다리의 위치를 캡처합니다. 그런 다음 해당 이미지도 내보냅니다. 마지막으로 PowerPoint와 같은 슬라이드 준비 소프트웨어를 사용하여 밴드와 사다리 이미지를 정렬하여 하나의 이미지를 형성합니다.
이 이미지는 흉선세포 세포에서 단백질 c-myc의 면역침전에 대한 웨스턴 블롯 결과를 보여줍니다. 왼쪽에서 오른쪽으로 레인은 isotype control, c-myc IP 및 pre-cleared lysate 입력을 나타냅니다. 맨 오른쪽의 차선은 분자량 사다리의 병합된 이미지입니다. 약 25 킬로달톤의 강한 띠는 경쇄에서 온 것이고 50 킬로달톤의 강한 띠는 결합 항체의 중쇄에서 온 것이며 IP 또는 샘플에 비특이적입니다. C-myc는 서쪽 얼룩에서 약 67 킬로달톤을 달리며 일반적으로 75 킬로달톤 사다리 띠 바로 아래에서 볼 수 있습니다. 이 블롯에서 c-myc 띠는 두 번째 레인에서 볼 수 있지만 첫 번째 레인에는 없는데, 이는 IP 항체가 c-myc를 성공적으로 끌어내렸음을 나타냅니다. pre-cleared lysate lane에는 눈에 보이는 띠가 없는데, 이는 이 단백질이 내인성 발현 수준이 낮다는 것을 시사합니다.
위에 자세히 설명된 절차의 결과는 그림 2에 나와 있습니다. 왼쪽에서 오른쪽으로, 차선에는 대조군(isotype), 시험 군(c-myc), 미리 지워진 용해(lysate), 분자량 사다리(사다리)가 포함됩니다. 25 kDa 래더 밴드가 표시됩니다. ~25 kDa 및 50 kDa의 두 개의 눈에 띄는 밴드는 결합 항체의 빛과 무거운 사슬이며, 각각 IP 또는 샘플에 특이적이지 않습니다. c-myc 단백질은 서양 얼룩에서 약 67kDa를 실행하고 일반적으로 바로 아래 볼 수 있습니다 75 kDa 사다리 밴드. 이 블롯에서 c-myc 대역은 두 번째 차선에서 볼 수 있지만 첫 번째 차선에는 존재하지 만 IP 항체가 성공적으로 c-myc를 끌어 내렸다는 것을 나타냅니다. 미리 지워진 용해 차선에는 눈에 보이는 밴드가 없기 때문에 이 단백질은 내인성 발현 수준이 낮다는 것을 시사합니다.
요컨대, 면역 침전은 항체를 이용한 특정 단백질의 분리이다. 이 예에서, 면역 침전의 결과는 순도를 평가하기 위해 서양 블롯에 의해 분석되었다. 고립 된 단백질은 단백질 구조를 위한 NMR, 아미노산 서열에 대한 질량 분광법 또는 효소 특성화를 위한 체외 분석 등 여러 가지 응용 분야에서 사용될 수 있습니다. IP는 또한 단백질의 상호 작용 파트너를 특성화할 수 있습니다. 예를 들면, 격리를 따르는, DNA 또는 RNA는 순서를 위해 격리될 수 있었습니다. 공동 면역 전성은 단백질 단백질 상호 작용을 평가합니다. 표적 단백질이 IP 도중 아래로 당길 때, 상호 작용하는 단백질은 또한 아래로 당길 수 있습니다. 이러한 상호 작용 파트너는 질량 분석 및 서양 얼룩에 의해 평가 될 수있다. 면역 강수량은 단백질 생물학을 연구하기위한 강력한 기술입니다.
Chapters in this video
0:01
Concepts
3:25
Immunoprecipitation Using Protein A/G Plus Agarose Beads
8:18
IP Verification Through Western Blot Analysis
12:36
Results
Videos from this collection: