Eukaryotic cells use an integrated network of glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation to meet the majority of their energy demands and to provide intermediates required for cell growth and proliferation. These pathways start with the intracellular trapping of free glucose in the form of glucose-6-phosphate, which subsequently gets processed into pyruvate. Pyruvate is either reduced to lactate or transported into the mitochondria, where it forms acetyl coenzyme A (CoA). Acetyl CoA then enters into the TCA cycle. High-energy intermediates of the TCA cycle propel the movement of electrons in the electron transport chain (ETC), which in turn expels H+ from the mitochondrial matrix to generate a H+ gradient across the inner mitochondrial membrane. Oxygen acts as the final electron acceptor, and H+ return back to the mitochondrial matrix through the FO/F1 complex, where their potential energy is used to generate ATP (Figure 1A).
The working principle of the extracellular flux assay depends on interfering with glycolysis and oxidative phosphorylation at specific points, and assessing the resulting effects. For this purpose, we used glucose to propagate glycolysis in starved cells, and 2-deoxyglucose (2-DG), which is converted to 2-deoxyglucose-6-phosphate, a competitive inhibitor of phosphoglucoisomerase23, by hexokinase, in order to block glycolysis. Rotenone (a complex I-specific inhibitor of the ETC), antimycin A (a complex III-specific inhibitor of the ETC), oligomycin (inhibitor of ATP synthase24), and the uncoupling agent 2,4-dinotrophenol (DNP)25 were used to intervene specific events related to electron transport, proton gradient and ATP synthesis (Figure 1A). Instead of 2,4-DNP, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) can also be used, but further optimization may be required. In either case, uncouplers should always be titrated for a specific cell type before use to determine the optimal concentration necessary.
The glycolysis stress test (Figure 1B) starts with a baseline measurement of the extracellular acidification rate (ECAR) in the previously starved cells. Since these cells are at their basal minimum, and can therefore be practically considered as non-glycolytic, the ECAR measured at this point is referred to as the non-glycolytic acidification. This acidification likely corresponds to respiratory CO2 generated in the TCA cycle being converted to HCO3– and H+. This is followed by the injection of glucose to activate glycolysis, which presents as an increase in the extracellular acidification due to the formation of lactate. This increase represents the normal rate of glycolysis. The cells are then challenged with the injection of oligomycin, which blocks the generation of ATP through oxidative phosphorylation. Cells respond to this dramatic decrease in ATP production by activating glycolysis to its maximum level, and that results in a secondary increase in the ECAR level (glycolytic reserve). The test is terminated by total inhibition of glycolysis using the glucose analog 2-DG, which returns the ECAR to its non-glycolytic level. Interestingly, it has been proposed that, contrary to the manufacturer's recommendations, maximal glycolysis is not necessarily attained by the injection of oligomycin26. In cells with a high glycolytic capacity, if there is not a significant increase in ATP demand, glycolysis may be perfectly able to cope with the loss of mitochondrial ATP without it needing to be up-regulated. The glycolytic rate with oligomycin could be surpassed by adding respiratory inhibitors, such as rotenone and myxothiazol, which provide a few benefits over oligomycin: 1) They increase the ATP demand, as they cause the reversal of ATP Synthase, with ATP hydrolysis, to pump protons in an attempt to recover the mitochondrial membrane potential26; 2) They prevent respiratory acidification of the medium, which can confound the ECAR results (see below). Other ways to increase ATP demand include the addition of compounds that stimulate the hydrolysis of ATP by the plasma membrane ATPases26. All this should be considered carefully by researchers when planning a glycolysis stress test.
The mitochondrial stress test (Figure 1C) starts with a baseline measurement of the oxygen consumption rate (OCR) in non-starved cells. This is followed by the injection of oligomycin, which inhibits the return of protons through the FO/F1 complex and thus rapidly hyperpolarizes the mitochondrial membrane. Hyperpolarization prevents further proton pumping through respiratory complexes, and the respiratory rate decreases. The remaining respiration is called proton leak, which represents the flow of protons through lipids or other channels. This hyperpolarized state is rapidly reversed by the addition of the uncoupling agent 2,4-DNP, which acts as a proton ionophore. In response, cells try to recover the membrane potential in a futile attempt by increasing the electron transport rate to its maximum, and this in turn increases the OCR. Finally, with the addition of two ETC inhibitors (antimycin A and rotenone), mitochondrial respiration completely stops and OCR decreases to its lowest level. At this level, oxygen consumption is not due to mitochondrial activity (non-mitochondrial). The difference in the OCR generated by these inhibitors is called the maximum mitochondrial respiration, which is the sum of the baseline respiration and the spare capacity.
B and T cell isolations yield highly viable pure lymphocyte populations.
B cells and naïve CD4+ T cells were isolated as outlined in Section 4 of the protocol, and splenocytes were simply obtained by lysing the red blood cells as described in Section 3.3.
The sensitivity of cell type-specific metabolic assays depends on the viability and the purity of the starting cell population. Therefore, in order to verify viability of the isolated mouse T cells, splenocytes, and B cells, and the purity of T and B cells, small aliquots of cells were stained for flow cytometric analysis (Figure 2). In the forward scatter-area vs. side scatter-area (FSC-A vs. SSC-A) plot, lymphocytes were gated, and within this gate, the population along the diagonal in the forward scatter-height (FSC-H) vs. FSC-A plot were determined as the singlets. Within the singlet population, viability was measured by gating the cells that stained negative for the live/dead marker (Figure 2A); T cell viability was 97.9%, splenocyte viability was 92%, and B cell viability was 94%. B cell purity, as measured by the B220+ CD19+ population26, was 99%, while CD4+ T cell purity, as measured by the CD44– CD4+ population27, was 98.3% (Figure 2B).
Protein concentration of the cell lysate can be used as a direct indicator of plated cell number.
Isolated lymphocytes and splenocytes were plated in an adhesive-coated 96-well assay plate at 5 x 105 cells/well, 2.5 x 105 cells/well, 1.25 x 105 cells/well, and 0.625 x 105 cells/well. The confluence at each plating density of the three cell types was visualized under the light microscope (Figure 3A). As expected, confluence correlated with the initial plating densities. Upon completion of the extracellular flux assay, plated cells were lysed and their protein concentrations were quantified using the BCA assay. For all cell types, lysate protein concentrations were shown to be linearly correlated with the initial plating densities (Figure 3B), which confirms that lysate protein concentrations can be used as an accurate measure for the normalization of cell numbers when interpreting the extracellular flux data. The plating densities used in this experiment have been optimized for naïve, unstimulated lymphocytes. If stimulated or previously cultured cells are to be used, further optimization may be required.
Mitochondrial and glycolytic stress assays are dependent on plated cell number.
The OCR was measured for each cell type and plating density in the extracellular flux analyzer. As expected, higher cell numbers have a higher measured OCR, as well as more dramatic responses to oligomycin, 2,4-DNP, and antimycin A/rotenone (Figure 4A). Standardizing OCR measurements to each sample's protein concentration reveals that in general, larger numbers of cells lead to more accurate OCR measurements. Plating at 5 and 2.5 x 105 cells/well resulted in similar normalized OCR measurements in the T cells and splenocytes, and 5 and 1.25 x 105 cells/well resulted similar normalized OCR measurements in B cells (Figure 4B). The slight differences in the normalized B cell OCR measurements might be an indirect indication that B cells perform better at 2.5 x 105 cells/well compared to other cell densities. By all measures, 0.625 x 105 cells/well gave suboptimal results, demonstrating that this plating density is insufficient. Baseline respiration, proton leak, maximum mitochondrial respiration, non-mitochondrial respiration, and ATP production linearly correlated with plating densities for all cell types (Figure 4C). Additionally, most of the baseline respiration is used towards synthesizing ATP, as indicated by the low proton leak in the three cell types. While the primary focus of the mitochondrial stress experiment is to measure changes in OCR, the ECAR is still useful to record to ensure that the assay was successfully carried out. Similar to the OCR, the two higher plating densities resulted in higher ECAR and more dramatic responses to oligomycin, 2,4-DNP, and antimycin A/rotenone (Figure 4D). The dramatic changes in ECAR upon addition of 2,4-DNP, and antimycin A/rotenone may be due to changes in respiration in addition to changes in glycolysis, since CO2 generated in the TCA cycle is converted to HCO3 – and H+. This issue has been recently addressed, and there exists a simple method for correcting the total extracellular acidification signal using oxygen consumption data, to obtain the real glycolytic rate12,29.
The glycolysis stress assay was most successful at the highest plating density (Figure 5A, B). In all cell types, the 5 x 105 cells/well samples had the greatest changes in ECAR after the addition of glucose, oligomycin, and 2-DG (Figure 5B). Additionally, normalizing to protein concentration demonstrated that for all cell types, the 5 x 105 cells/well samples yielded optimal results, while lower concentrations-especially 0.625 x 105 cells/well-did not display a robust glycolytic reserve capacity. Non-glycolytic acidification, glycolysis, and apparent glycolytic capacity linearly correlated with plating densities for all cell types (Figure 5C). For the glycolysis stress experiment, the OCR graph shows that glucose slightly stimulates mitochondrial respiration, which is then inhibited by oligomycin treatment; the addition of 2-DG did not affect the OCR (Figure 5D). The OCR graph can be used as a further indicator that the glycolysis stress experiment was successfully carried out. Also, the OCR graph can be used to correct the ECAR graph, if required, as explained above in the case of the mitochondrial stress test12,29.

Figure 1: Outline of extracellular flux assays. (A) Illustration of glycolysis (left) and oxidative phosphorylation (right) showing the action of the metabolic drugs used in the extracellular flux assays. (B) Schematic of the extracellular acidification rate (ECAR) graph; schematic of the oxygen consumption rate (OCR) graph (C). Please click here to view a larger version of this figure.

Figure 2: Stepwise gating of T cells, B cells, and splenocytes to determine viability and purity. Flow cytometry plots showing the viability of T cells, splenocytes, and B cells (A); and purity of T and B cells (B). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
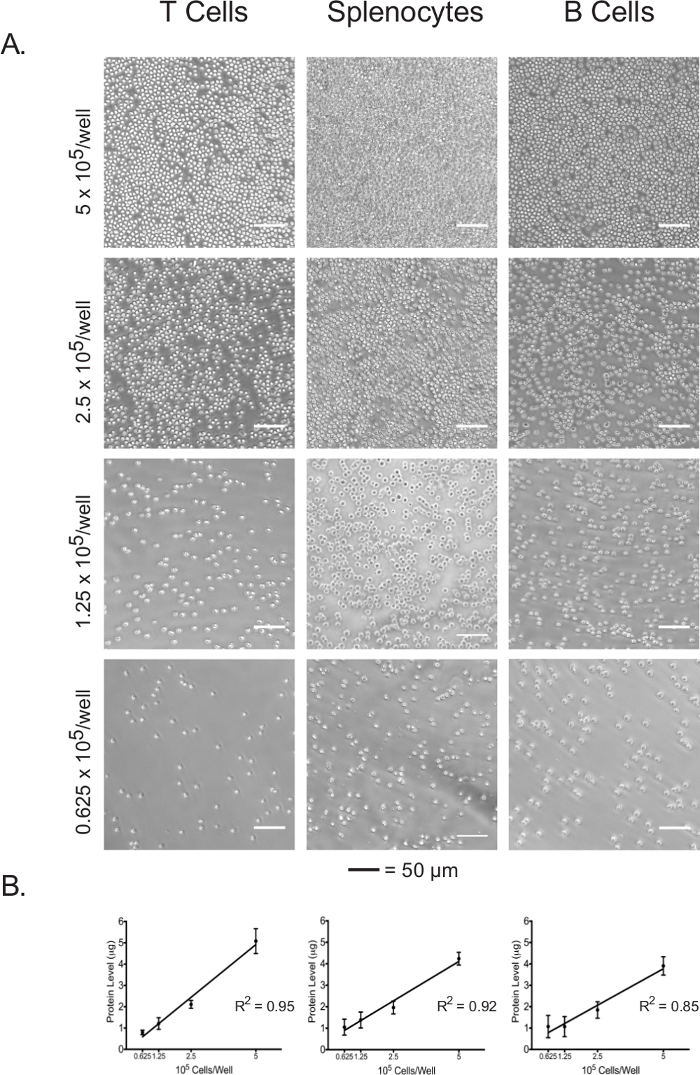
Figure 3: Cell confluence correlates with the lysate protein concentrations of each cell type at different plating densities. (A) Light micrographs of cells in assay plate wells at plating densities ranging from 5 x 105 cells/well to 0.625 x 105 cells/well. Scale bars denote 50 µm. (B) Lysate protein concentrations at different plating densities, as measured by the BCA assay. Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.

Figure 4: Mitochondrial stress assay. Raw (A) and standardized (B) OCR for each cell type and plating density are shown. (C) Cell number-dependent changes in the levels of baseline, maximum mitochondrial and non-mitochondrial respiration, as well as the oxygen consumptions linked to proton leak or ATP production, are shown for each cell types. (D) Raw ECAR values obtained from the mitochondrial stress tests are shown. Each data point represents the mean of 7-8 wells with standard deviation. Labeled arrows denote injections of oligomycin (O), 2,4-DNP (D), and rotenone/antimycin A (R/A). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.

Figure 5: Glycolytic stress assay. Raw (A) and standardized (B) ECAR for each cell type and plating density are shown. (C) Cell number-dependent changes in ECAR for non-glycolytic acidification, glucose-induced glycolysis and total glycolytic capacity are shown. (D) Raw OCR values obtained from the glycolytic stress tests are shown. Each data point represents the mean of 7-8 wells with standard deviation. Labeled arrows denote injections of glucose (G), oligomycin (O), and 2-deoxyglucose (2-DG). Results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
