

Summary
La combinaison de l'immunoprécipitation de la chromatine et le séquençage ultra-haut débit (chip-seq) permet d'identifier et de cartographier les interactions protéine-ADN dans une lignée de tissu ou de cellule donné. Décrit est de savoir comment générer un modèle de puce de haute qualité pour le séquençage ultérieur, en utilisant l'expérience avec le facteur de transcription TCF7L2 comme un exemple.
Abstract
ChIP-séquençage (ChIP-seq) méthodes offrent directement couverture du génome entier, où la combinaison immunoprécipitation de la chromatine (ChIP) et le séquençage massivement parallèle peut être utilisé pour identifier le répertoire de séquences d'ADN de mammifères liés par des facteurs de transcription in vivo. "La nouvelle génération" technologies de séquençage du génome fournissent 1-2 ordres de grandeur dans l'augmentation de la quantité de séquence qui peut être rentable généré plus anciennes technologies permettant ainsi des méthodes ChIP-Seq pour fournir directement la couverture du génome entier pour le profilage efficace des mammifères interactions protéine-ADN.
Pour les approches ChIP-Seq succès, il faut générer une matrice d'ADN de Morceau de qualité afin d'obtenir les meilleurs résultats de séquençage. La description est basée sur l'expérience avec le produit protéique du gène le plus fortement impliquée dans la pathogenèse du diabète de type 2, à savoir le facteur de transcription facteur de transcription 7-like 2 (TCF7L2). Ce facteur a également été impliqué dans divers cancers.
Décrit est de savoir comment générer une matrice d'ADN de puce de haute qualité issu de la lignée de cellules de carcinome colorectal, HCT116, afin de construire une carte à haute résolution par séquençage de déterminer les gènes liés par TCF7L2, donner un nouvel aperçu de son rôle clé dans la pathogenèse des traits complexes.
Introduction
Depuis de nombreuses années, il ya eu un besoin non satisfait pour identifier l'ensemble des gènes liés et réglementée par une protéine échelle du génome entier, en particulier compte tenu de ceux de la classe de facteur de transcription.
Odom et al. 1 utilisé immunoprécipitation de la chromatine (ChIP) combiné avec des puces de promoteurs afin d'identifier systématiquement les gènes occupées par des régulateurs de transcription pré-spécifiés dans le foie humain et îlots pancréatiques. Par la suite, Johnson et al. 2 mis au point un test d'immunoprécipitation de la chromatine à grande échelle basée sur le séquençage direct ultra haut débit ADN (ChIP-seq) afin de cartographier en détail les interactions protéine-ADN dans l'ensemble de génomes de mammifères. En cas de test, ils ont cartographié in vivo la liaison du facteur de silencieux neurones restrictive (CRSN) à 1946 endroits dans le génome humain. Les données affichées résolution forte de position de reliure (+ 50 paires de bases), ce qui a facilité à la fois le iconsolation de motifs et de l'identification des motifs CRSN contraignantes. Ces données ChIP-Seq également eu une grande sensibilité et de la spécificité et de la confiance statistique (p <10 -4), propriétés qui sont importantes pour déduire de nouvelles interactions candidats.
Robertson et al. 3 également utilisé ChIP-seq afin de cartographier STAT1 cibles dans l'interféron γ-(IFN-γ)-stimulés et non stimulés cellules HeLa S3 humains in vivo. Par Chip-Seq, avec 15,1 et 12,9 millions de séquence cartographiée unique lit, et un taux de fausses découvertes estimée à moins de 0.001, ils ont identifié 41 582 et 11 004 régions STAT1-putatifs de liaison dans les cellules stimulées et non stimulées, respectivement. Sur les 34 loci connus pour contenir l'interféron STAT1 sensible liaison 4-8 sites ChIP-seq trouvé 24 (71%). Objectifs ChIP-Seq ont été enrichies en séquences similaires à connaître STAT1 motifs de liaison. Les comparaisons avec les données de la puce deux-PCR existants définit suggéréque la sensibilité ChIP-seq se situait entre 70% et 92% et la spécificité était d'au moins 95%. En outre, il était clair que ChIP-seq offre à la fois une faible complexité analytique et une sensibilité qui augmente avec la profondeur de séquençage.
Alors que les technologies de séquençage tel, Génome "nouvelle génération" offrent 1-2 ordres de grandeur dans l'augmentation de la quantité de séquence qui peut être rentable généré plus anciennes technologies 9. Méthodes ChIP-Seq fournissent donc directement couverture du génome entier pour le profilage efficace des interactions protéine-ADN de mammifères 3.
En 2006, une forte association des variants du facteur de transcription 7-like 2 (TCF7L2) gène du diabète de type 2 a été découvert 10. D'autres chercheurs ont déjà répliqué indépendamment de cette constatation dans différentes ethnies et, curieusement, depuis les premières études d'association génomique échelle du diabète de type 2 publiés dans Nature 11,12 Science 13-15 et ailleurs 16,17, la plus forte association était en effet avec TCF7L2, ce qui est maintenant considéré comme le résultat génétique le plus important dans le diabète de type 2 à ce jour 18-20. En outre, TCF7L2 a été liée à un risque de cancer 21,22, en effet, ce lien est devenu plus évident lorsque le locus 8q24 révélé par Génome larges études d'association d'un certain nombre de cancers, y compris les carcinomes colorectaux, s'est révélée être en raison d'une extrême amont TCF7L2 élément contraignant de conduire la transcription MYC 23,24. En tant que tel, il ya un grand intérêt dans la détermination des gènes en aval régulés par ce facteur de transcription clé.
Basé sur l'expérience avec TCF7L2 comme un exemple de la méthodologie, ce document décrit comment générer une matrice d'ADN de Morceau de qualité. Puce a été effectuée de la lignée cellulaire de carcinome colorectal, HCT116, pour le séquençage ultérieur afin de construire un haut-résolcarte ution des gènes liés par TCF7L2 25 dans un effort pour donner un meilleur aperçu de son rôle clé dans la pathogenèse de traits complexes.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Cross-link chromatine
- Cultiver les cellules dans des boîtes de culture cellulaire 100x20mm. La quantité de cellules peut varier de 1 à 10 millions de cellules par boîte selon le type de cellule. Environ 2 millions de cellules est suffisant pour une immunoprécipitation.
- Cellules Cross-Link 1% de formaldéhyde pendant 10 min à température ambiante avec bascule occasionnelle.
- Étancher la réticulation en ajoutant une concentration finale de 125 mM de glycine et incuber pendant 5 minutes à température ambiante.
- Laver les cellules avec du phosphate 1X saline tamponnée (PBS) deux fois, décanter PBS, puis ajoutez 0,2 ml de PBS.
- Récolte des cellules avec un grattoir en plastique cellulaire dans un tube de centrifugation.
- Isoler les cellules à 2000 rpm pendant 5 min à 4 ° C.
- Aspirer le surnageant. Remettre les cellules dans un tampon de lyse SDS (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8,1) pour lysat cellulaire ou les garder comme une pastille pour l'extraction nucléaire.
- Les cellules peuvent être sauvegardés à -80 ° C ou l'on peut procéder immediatEly avec ultrasons.
2. Préparer noyaux (Passez à l'étape 3.5 pour lysat de cellules entières)
- Supplément tampon de lyse cellulaire (5 mm tuyaux pH 8,0, 85 mM KCl, 0,5% de NP-40) avec 1X inhibiteur de la protéinase chaque expérience.
- Reprendre décongelé culot cellulaire dans environ 10 fois le volume de granulés avec le tampon de lyse cellulaire.
- DOUNCE-homogénéiser 10 fois avec un pilon puis incuber sur de la glace pendant 10 min.
- Centrifuger à 4000 rpm pendant 5 min à 4 ° C, éliminer le surnageant et sauver culot nucléaire.
3. Sonication *
- Réchauffez-vous SDS tampon de lyse et le montant du supplément de tampon pour être utilisé avec l'inhibiteur de la protéinase.
- Remettre en suspension culot nucléaire en SDS Lysis Buffer (environ 0,5 ml de tampon par 1-10000000 cellules)
- Incuber sur de la glace pendant 10 min.
- Ajouter 0,5 ml d'aliquotes d'échantillons à des microtubes.
- Soniquer sur de la glace à l'aide Misonix ultrasons avec 30 sec sur sec et 45off avec un réglage d'amplitude de 2. Nombre de cycles de taille idéale de fragment peut être déterminée en premier essayer différents nombres de cycles (ex. 2, 4, 8, 12, 16 et 20 cycles ou plus). Une autre marque de sonicator peut être utilisé, cependant, les conditions peuvent varier. L'expérimentation avec le nombre de cycles et la quantité de temps sur et en dehors doivent être effectuées afin de déterminer les conditions idéales.
- Recueillir 20 ul de chaque échantillon pour vérifier les résultats de sonication et faire quantification. Le reste de l'échantillon peut être conservé à -80 ° C.
- Diluer le 20 ul d'échantillon en ajoutant 30 ul de 0,1 X TE Buffer.
- Traiter l'échantillon avec 1 pi de RNase A à 37 ° C pendant 1 heure puis ajouter 1 l de protéinase K et incuber à 62 ° C pendant 2 heures.
- Exécuter 20 pl de l'échantillon sur un gel d'agarose à 2%.
- Purifier le montant restant de l'échantillon avec QIAquick PCR purification kit puis quantifier en utilisant un spectrophotomètre NanoDrop.
* Pour Chi nativeP, microccocal digestion à la nucléase peut être utilisé alternativement pour cisailler l'ADN.
4. Bloquer Agarose perles *
- Si les perles sont déjà bloqués, passez à l'étape 5.1.
- Utilisez une protéine ou protéine G agarose. Pour 5 immunoprécipitat (IPS), utiliser 600 ul de 50% Billes en suspension (300 pi perle granulés)
- Pour laver les perles, les faire tourner vers le bas à 800 rpm pendant 1 min à 4 ° C et éliminer le surnageant. Ajouter un peu plus de 2 ml ChIP Dilution Buffer (0,01% SDS, 1,2 mM EDTA, NaCl 167 mM, 1,1% de Triton X-100, Tris-HCl 16,7 mM pH 8.1) et mélanger en retournant lentement le tube 10X. Tournez à nouveau à 800 rpm pendant 1 min à 4 ° C et éliminer le surnageant. Répétez les lave 2 fois plus.
- Bloc billes en faisant tourner à 4 ° C pendant une nuit dans une solution de blocage. Reportez-vous au tableau 1 pour la recette de la solution de blocage.
5. Chromatine pré-claire
- Dégel ultrasons chromatine sur la glace.
- Isoler à 12000 rpm fou 10 min à 4 ° C puis mis sur la glace tout de suite pour éliminer le SDS (blanc boulette).
- Recueillir le surnageant, jetez granulés, et combiner des échantillons si nécessaire.
- Sortez les montants nécessaires pour l'expérience basée sur des calculs (1-10 ug de la chromatine par IP).
- Diluer la chromatine 10X tampon de dilution ChIP complété par un inhibiteur de protéinase.
- Ajouter 100 ul de billes bloqués par IP.
- Tournez à 4 ° C pendant 1 heure.
6. Immunoprécipitation
- Isoler les échantillons à 800 rpm pendant 1 min et transférer le surnageant dans un nouveau tube.
- Isoler surnageant à 800 rpm pendant 1 min et le transfert à un autre tube propre.
- Economisez 20 pi du surnageant, pour servir de contrôle d'entrée, à -20 ° C.
- Aliquote de la chromatine au nombre d'adresses IP à faire dans l'expérience.
- Ajouter 2 ug d'anticorps par 1-10 ug de la chromatine à chaque échantillon.
- Incuber pendant 4 ° C avec rotation.
- Ajouter 100 ul de billes bloqués pour chaque échantillon IP.
- Incuber pendant 1 heure à 4 ° C avec rotation.
- Pellet les perles en faisant tourner vers le bas à 800 rpm pendant 1 min et jeter autant de surnageant que possible.
- Lavez perles fois avec du tampon de lavage complexe immunitaire Low Salt. Ajouter 1 ml de tampon à chaque tube, tourner à température ambiante pendant 5-8 min; ralentit à 800 rpm pendant 1 min, puis éliminer le surnageant. Répétez laver une fois avec le tampon de haut sel complexe immun de lavage et un tampon de lavage complexe immunitaire LiCl et deux fois avec du tampon TE pour un total de 5 lavages (tableau 2).
7. Élution
- échantillons d'entrée de dégel du jour précédent à traiter avec les éluants.
- Faire Elution Buffer frais (tableau 3).
- Faites un mélange maître de suffisamment de tampon d'élution nécessaire pour IPs et les échantillons de contrôle d'entrée plus 1-2 échantillons supplémentaires.
- Ajouter 100 pi de tampon d'élution à chaque échantillon IP et incuber à température ambiante pendant 15 mdans la rotation.
- Isoler à 800 rpm pendant 1 min et ajouter le surnageant dans un nouveau tube.
- Ajouter un autre 100 pi de tampon d'élution à chaque tube de perles et incuber à température ambiante pendant 15 min avec rotation.
- Vortex pendant 15 secondes après incubation, centrifuger à 5000 rpm pendant 1 min, puis les combiner surnageant avec le surnageant de la première élution. (Assurez-vous qu'il n'y a pas laissés sur des billes dans les surnageants. Cas de doute, ralentit à nouveau surnageant à 5000 rpm pendant 1 minute et recueillir le surnageant dans un nouveau tube.
- Ajouter 180 ul de tampon d'élution à la 20 pl d'échantillons de contrôle d'entrée.
8. Inverser la Croix-link
- Pour les 200 pi de éluants et les contrôles d'entrée, ajouter 8 pi de 5 M de NaCl.
- tubes de sceller avec du Parafilm et incuber dans un bain d'eau à 65 ° C pendant la nuit.
9. Purification de l'ADN
- Traiter chaque échantillon avec 1 pi de RNase A pendant 1 heure à 37 ° C.
- Ajouter 4 μl d'EDTA 0,5 M, 8 pi 1M Tris-HCl, mélanger, puis ajouter 1 l de protéinase K à chaque échantillon et incuber à 45 ° C pendant 2 heures.
- Purifier les échantillons à l'aide QIAquick PCR purification kit. Les échantillons peuvent être enregistrés à -20 ° C et chèque PCR peut être fait à une date ultérieure.
* Alternativement, les billes magnétiques ChIP-grade peuvent être utilisés à la place d'agarose pour la partie immunoprécipitation.
10. Chèque PCR
- Pour le contrôle PCR, utiliser des amorces pour les régions connues pour être lié par la protéine d'intérêt. En outre, utiliser des amorces pour les régions non contraignants que les contrôles négatifs.
- Mélanger les réactifs pour la réaction. Diluer l'échantillon d'entrée à 1:100 (tableau 3).
- Exécuter réaction. Programme PCR:
Etape 1: 94 ° C 3 min
Étape 2: 94 ° C 20 sec
59 ° C 30 sec
72 ° C 30 sec
(Répétez l'étape 2 pour au moins 30 cycles)
Etape 3: 72 ° C 2 min
- Exécuter des échantillons sur gel agarose 1%.
- L'enrichissement peut aussi être déterminée de manière quantitative par PCR en temps réel.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Une fois la chromatine a été traité par ultrasons et ont été traités à la RNase et protéinase, les échantillons analysés sur le gel à 2% d'agarose doivent présenter un frottis avec la majeure partie de l'ADN à la taille désirée. Si plusieurs cycles différents sont testés, une diminution progressive de la taille doit être considéré comme le nombre de cycles de hausse (figure 2).
Après avoir terminé la partie d'immunoprécipitation du protocole de l'enrichissement peut soit être vérifiée par PCR ou PCR en temps réel. Pour les échantillons de PCR sur un gel d'agarose il devrait y avoir des bandes dans l'entrée et à puce (en utilisant un anticorps pour la protéine d'intérêt, ce qui est TCF7L2 dans ce cas) les voies de l'échantillon et rien ou tout au plus, une bande très faible (bruit de fond) dans le IgG (négatif) voie de commande pour la région de liaison positive. Pour la région de liaison négative il devrait être très faible ou pas de bande pour le contrôle IgG et voies de puce. Il devrait y avoir une bande sur la voie d'entrée (Figure 3).
La figure 4 montre les mêmes échantillons examinés par PCR en temps réel. Comme dans la figure précédente, il devrait y avoir un enrichissement pli important de la région de liaison positif pour l'échantillon ChIP sur le contrôle IgG. En outre, il devrait y avoir très peu d'enrichissement, le cas échéant, vu dans la région de liaison négative.

Figure 1. Schéma du processus de puce. Cliquez ici pour agrandir la figure .
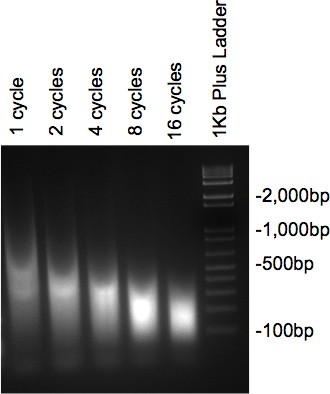
Figure 2. Gel vérifier des ultrasons de l'ADN.

Figure 3. PCR Vérifiez la puce.
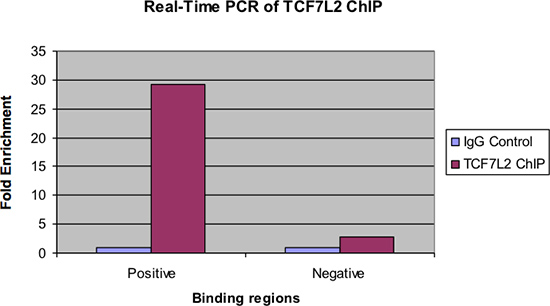
Figure 4. PCR en temps réel des TCF7L2 puce.
| Réactif | Volume |
| Perle pastille | 300 pi |
| BSA (50 mg / ml) | 30 pl |
| 100X inhibiteur de la protéinase | 10 pl |
| Dilution Buffer ChIP | 660 ul |
| Total | 1000 pi |
Tableau 1. Recette pour bloquer agarose.
| Tampon | Composants |
| Faible teneur en sel immunitaire tampon de lavage Complex | SDS 0,1% 1% Triton X-100 2 mM EDTA Tris-HCl 20 mM pH 8,1 NaCl 150 mM |
| High Salt immunitaire tampon de lavage Complex | SDS 0,1% 1% Triton X-100 2 mM EDTA Tris-HCl 20 mM pH 8,1 NaCl 500 mM |
| LiCl Immune tampon de lavage Complex | 0,25 M de LiCl 1% de NP-40 1% Deoxycholate EDTA 1 mM Tris-HCl 10 mM pH 8,1 |
| TE Buffer | Tris-HCl 10 mM pH 8,1 EDTA 1 mM, pH 8,0 |
Tableau 2. tampons de lavage de puce.
| Réactif | Volume |
| 10 pl | |
| 1 M NaHCO3 | 20 pl |
| H 2 O | 170 ul |
Tableau 3. tampon d'élution pour une IP.
| Réactif | 50 Réaction ul | 20 Réaction ul |
| Eau | 27 ul | 10,8 pi |
| Tampon de réaction PCR 5X | 10 pl | 4 pl |
| MgCl2 | 4 pl | 1,6 pl |
| dNTP (10 mM) | 1 pl | 0,4 pl |
| mélange d'amorces (5 uM chacun) | 2 pl | 0,8 pl |
| Taq (Promega Hotstart) | 1 <ul/ Td> | 0,4 pl |
| ADN ChIP | 5 pl | 2 pl |
Tableau 4. Les volumes de réaction de PCR.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Il est désormais possible d'effectuer l'ensemble du génome profil de l'association des interactions protéine-ADN en utilisant ChIP-seq, comme cela a été récemment démontrée avec d'autres facteurs de transcription 2,3. La clé d'un résultat de séquençage réussie est la génération d'un modèle chromatine de l'ADN d'immunoprécipitation de haute qualité.
Une fois la matrice d'ADN a été généré et déterminé à être enrichi de manière adéquate, on peut alors prendre en prévision de la bibliothèque pour le séquençage ultérieur. Par exemple, on peut utiliser le protocole de séquençage bibliothèque fournie par le fournisseur, Illumina. La sélection de la taille de cette bibliothèque peut être effectuée par électrophorèse sur gel et l'excision subséquente et la purification de l'ADN dans le ~ 200 - à l'echelle 700 pb. La réduction de la taille et de la réduction de la gamme de taille d'ADN prélevés dans la purification de gel est destiné à améliorer la résolution de position de ChIP-seq. En enrichissant pour les petits morceaux d'ADN d'entrée liés au facteur de intEREST, on pourrait s'attendre à ce que l'emplacement du site gagnera résolution. Sélection de la taille serrée améliore également la taille uniformité des colonies moléculaires produites sur la plateforme Illumina. Une telle uniformité de taille des colonies augmente également le nombre de lecture effective obtenue. Entrée Shorter taille de l'ADN produit aussi des colonies les plus robustes sur la plate-forme Illumina, ce qui peut signifier que des morceaux d'ADN courtes dans toute distribution de l'échantillon d'entrée donné seront représentés de manière plus efficace dans la sortie de séquence finale que sont des morceaux d'entrée plus de la même distribution.
Les approches bioinformatiques pour l'analyse de la séquence "nouvelle génération" ne cessent d'évoluer, avec de nombreux vendeurs font leur logiciel open-source pour plus de raffinement. On peut transformer les lectures qui correspondent aux emplacements génomiques uniques dans un fragment profil de recouvrement de l'ADN. Pics significatifs peuvent être identifiés par des profils de threshholding à une hauteur équivalente à un taux de fausses découvertes estimé. La position spmatrices de fréquences SPÉCIFIQUE dérivés de ce travail peut être utilisé pour identifier et localiser les sites de liaison de l'ADN dans le génome humain pour un facteur donné.
Mais il faut être prudent en ce qui concerne les facteurs qui l'on souhaite étudier avec ChIP-seq. Avant d'entreprendre une telle étude doit évaluer si un un anticorps est disponible sur le marché qui est utilisable dans le cadre de la puce, comme un pauvre anticorps peut avoir des effets très néfastes sur les résultats expérimentaux de chacun. En outre, on doit considérer s'il ya des isoformes d'épissage de la protéine à l'étude, en effet, TCF7L2 est connu pour avoir de nombreuses isoformes nous étions donc particulièrement prudent dans le choix d'un anticorps qui se lie aux acides aminés toujours présents dans tous les principaux isoformes de ce facteur de transcription 25.
En résumé, la combinaison d'immunoprécipitation de la chromatine et le séquençage ultra-haut débit (ChIP-seq) permet d'identifier et de cartographier les interactions protéine-ADN dans un tissu ou c donnéligne d'ell. Nous avons décrit comment générer un modèle de jeton de haute qualité pour séquençage ultérieur.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs déclarent qu'ils n'ont aucun intérêt financier concurrents.
Acknowledgments
Le travail est soutenu par un prix de l'Institut de développement de l'Hôpital pour enfants de Philadelphie.
Materials
| Name | Company | Catalog Number | Comments |
| QIAquick PCR Purification Kit | Qiagen | 28104 | |
| EZ-ChIP Kit | Millipore | 17-371 | |
| GoTaq Hot Start Polymerase | Promega | M5001 | |
| Misonix Sonicator | Qsonica | XL-2000 | |
| NanoDrop 1000 Spectrophotometer | Thermo-Scientific | ||
| Positive control primer sequences (TCF7L2-1) Forward- 5'-TCGCCCTGTCAATAATCTCC-3' Reverse- 5'-GCTCACCTCCTGTATCTTCG-3' Negative control primer sequences (CTRL-1) Forward-5'-ATGTGGTGTGGCTGTGATGGGAAC-3' Reverse- 5'-CGAGCAATCGGTAAATAGGTCTGG-3' |
|||
References
- Odom, D. T., et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 303, 1378-1381 (2004).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316, 1497-1502 (2007).
- Robertson, G., et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature Methods. 4, 651-657 (2007).
- Reich, N. C., Liu, L.
- Lodige, I., et al. Nuclear export determines the cytokine sensitivity of STAT transcription factors. The Journal of Biological Chemistry. 280, 43087-43099 (2005).
- Schroder, K., Sweet, M. J., Hume, D. A. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology. 211, 511-524 (2006).
- Vinkemeier, U. Getting the message across, STAT! Design principles of a molecular signaling circuit. The Journal of Cell Biology. 167, 197-201 (2004).
- Brierley, M. M., Fish, E. N.
- Bentley, D. R.
- Grant, S. F., et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics. 38, 320-323 (2006).
- Sladek, R., et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 445, 881-885 (2007).
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447, 661-678 (2007).
- Saxena, R., et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 316, 1331-1336 (2007).
- Zeggini, E., et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 316, 1336-1341 (2007).
- Scott, L. J., et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 316, 1341-1345 (2007).
- Steinthorsdottir, V., et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nature Genetics. 39, 770-775 (2007).
- Salonen, J. T., et al. Type 2 Diabetes Whole-Genome Association Study in Four Populations: The DiaGen Consortium. American Journal of Human Genetics. 81, 338-345 (2007).
- Zeggini, E., McCarthy, M. I. TCF7L2: the biggest story in diabetes genetics since HLA. Diabetologia. 50, 1-4 (2007).
- Weedon, M. N.
- Hattersley, A. T. Prime suspect: the TCF7L2 gene and type 2 diabetes risk. The Journal of Clinical Investigation. 117, 2077-2079 (2007).
- Yochum, G. S., et al. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 104, 3324-3329 (2007).
- Duval, A., Busson-Leconiat, M., Berger, R., Hamelin, R. Assignment of the TCF-4 gene (TCF7L2) to human chromosome band 10q25.3. Cytogenet. Cell Genet. 88, 264-265 (2000).
- Pomerantz, M. M., et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nature Genetics. 41, 882-884 (2009).
- Tuupanen, S., et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nature Genetics. 41, 885-890 (2009).
- Zhao, J., Schug, J., Li, M., Kaestner, K. H., Grant, S. F. Disease-associated loci are significantly over-represented among genes bound by transcription factor 7-like 2 (TCF7L2) in vivo. Diabetologia. 53, 2340-2346 (2010).
- Benjamini, Y., Yekutieli, D. Quantitative trait Loci analysis using the false discovery rate. Genetics. 171, 783-790 (2005).
