

Summary
クロマチン免疫沈降法と超高スループットシーケンシング(チップ-SEQ)の組み合わせは、所定の組織または細胞株におけるタンパク質-DNA相互作用を特定し、マッピングすることができます。概説としては、例えば、転写因子TCF7L2の経験を使用して、その後の配列決定のための高品質のChIPテンプレートを生成する方法である。
Abstract
クロマチン免疫沈降(ChIPの)を組み合わせて、超並列配列決定は、in vivoでの転写因子によって結合哺乳類のDNA配列のレパートリーを識別するために利用できる場所のChIP-シーケンシング(のChIP-seqの)メソッドを直接、全ゲノムカバレッジを提供する。 "次世代"ゲノムシーケンシング技術は、費用対効果的にこうして直接哺乳類の効果的なプロファイリングに全ゲノムカバレッジを提供するためのChIP-seqの方法を可能に古い技術の上に生成することができます順序の量で大きさの増加1-2受注を提供タンパク質-DNA相互作用。
成功したChIP-seqのアプローチの場合、人は最高のシークエンシング結果を得るために、高品質のチップDNAテンプレートを生成する必要があります。説明はすなわち最も強く、2型糖尿病の病因に関与する遺伝子のタンパク質産物の経験、転写因子の転写因子7のような2(TCF7L2に基づいています)。この因子はまた、種々の癌に関与している。
概説病因におけるその重要な役割にさらなる洞察を与えて、TCF7L2によって結合する遺伝子を決定するために配列決定を介して高解像度のマップを構築するために、HCT116、結腸直腸癌細胞株に由来する高品質のChIP DNAテンプレートを生成する方法である複雑な形質の。
Introduction
長年にわたり、特に所定のタンパク質のゲノムワイド、転写因子のクラスのものに拘束され、規制を受けた遺伝子のセットを識別するために満たされていない必要があった。
オドムら 1は、系統的にヒト肝及び膵島であらかじめ指定された転写調節因子によって占有された遺伝子を同定するためのプロモーターマイクロアレイと結合しクロマチン免疫沈降(チップ)を使用しました。その後、ジョンソンら 2総合的全体哺乳類ゲノムにわたるタンパク質-DNA相互作用をマッピングするために直接超高スループットDNAシークエンシング(チップ-SEQ)に基づく大規模なクロマチン免疫沈降アッセイを開発しました。テストケースとして、彼らは、生体内でヒトゲノムに1946の場所にニューロン制限サイレンサー因子(NRSF)の結合をマップしました。 iの両方を促進結合位置のシャープな解像度(+ 50塩基対)を表示されたデータ、モチーフとNRSF結合モチーフの同定ゾル化。これらのChIP-seqのデータはまた、高感度、特異性および統計的信頼度(P <10 -4)、新しい候補の相互作用を推定するために重要である特性を有していた。
ロバートソンら 3もでSTAT1ターゲットをマッピングするためにのChIP-seqのを使用したインターフェロン-γ(IFN-γ)で刺激し、 生体内でのヒトのHeLa S3細胞を刺激していない。のChIP-seqのことで、15.1と1290万一意にマッピングされたシーケンスが読み取り、0.001未満の推定偽発見率を使用して、彼らはそれぞれ、刺激し、刺激されていない細胞では41582と11004推定STAT1結合領域を同定した。 STAT1インターフェロン応答性結合部位の4-8を含むことが知られている34の遺伝子座のうち、チップseqが24(71%)を発見。のChIP-seqのターゲットは知らSTAT1結合モチーフに類似した配列に富んでいた。 2既存のChIP-PCRデータとの比較が示唆セットのChIP-seqの感度は70%と92%の特異性との間であったことは、少なくとも95%であった。さらに、それは、チップseqの深さを配列とともに増加低く、分析の複雑さと感度の両方を提供することが明らかになった。
このように、 "次世代の"ゲノムシーケンシング技術は、費用対効果的に古い技術9経由で生成することができます順序の量の大きさの増加の1-2注文を提供します。のChIP-seqの方法は、したがって、直接哺乳類タンパク質-DNA相互作用の3の効果的プロファイリングに全ゲノムカバレッジを提供。
2006年には、転写因子2 7-様(TCF7L2)2型糖尿病と遺伝子の変異の強い関連は10を発見されました。他の研究者は、すでに独立して自然 11,12年に出版、2型糖尿病の最初のゲノムワイド関連研究から、興味深いことに、さまざまな民族ではこの知見を複製しています科学 13-15と16,17アップ、最強協会はTCF7L2と確かにあったが、これは今では2型糖尿病の中で最も重要な遺伝子の発見は18-20をこれまでに考えられている。また、TCF7L2は癌リスク21,22にリンクされている;大腸癌を含む癌の数、のゲノムワイド関連研究によって明らかになった8q24の軌跡は、極端なアップストリームに起因することが示されたときに確かに、この接続がより明らかになりましたTCF7L2結合要素はMYC 23,24の転写を駆動する。このように、この重要な転写因子によって調節下流の遺伝子を決定することに大きな関心がある。
方法論の例としてTCF7L2と経験をもとに、本論文では、高品質のチップのDNAテンプレートを生成する方法を概説します。チップは高レゾールを構築するために、後続の配列決定のために、HCT116、結腸直腸癌細胞株において行った複雑な形質の病因におけるその重要な役割にさらなる洞察を得るための努力でTCF7L2 25により拘束遺伝子のutionマップ。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1。クロスリンククロマチン
- 100x20mm細胞培養皿で細胞を成長させる。細胞の量は、1〜細胞型に応じてディッシュあたり10万個の細胞の範囲とすることができる。約200万個の細胞は、一免疫沈降のために十分である。
- 時折揺らしながら室温で10分間、1%ホルムアルデヒドでクロスリンク細胞。
- 室温で5分間、125mMのグリシンとインキュベートの最終濃度を加えることによって架橋をクエンチ。
- 1Xリン酸で細胞を洗浄すると、二回デカントPBSを緩衝生理食塩水(PBS)、次にPBS 0.2 mlを加える。
- マイクロ遠心チューブにプラスチックセルスクレーパーで細胞を回収。
- 4で5分間2,000 rpmで、細胞をスピンダウン℃まで
- 上清を吸引。全細胞溶解用SDS溶解バッファーで再懸濁した細胞(1%SDS、10mMのEDTA、50mMのトリス-HCl pHは8.1)、またはそれらの核の抽出のためのペレットとして保つ。
- 細胞を-80℃で保存することができます°Cまたは1つはimmediatを進むことができます超音波処理とイーリー。
2。 (全細胞溶解のために3.5に進みます)核を準備
- 各実験では、1Xプロテイナーゼ阻害剤と細胞溶解バッファー(5mMのパイプpH8.0で、85のKCl、0.5%NP-40)を補う。
- 約10倍の細胞溶解バッファーでペレットボリュームで解凍細胞ペレットを再懸濁します。
- 乳棒で10回ダウンス·均質化し、10分間、氷上でインキュベートする。
- 4°Cで5分のために4,000 rpmで遠心し、上清を廃棄し、核ペレットを保存します。
3。超音波処理*
- SDS溶解バッファーとプロテイナーゼ阻害剤で使用するバッファの補充量をウォームアップ。
- SDS溶解バッファーで再懸濁し核ペレット(1-10万個の細胞あたりのバッファの約0.5ml)を
- 氷上で10分間インキュベートする。
- マイクロチューブにサンプル0.5mlのアリコートを追加します。
- 30秒で、45秒でMisonix超音波処理を用いたウェット、氷上で超音波処理オフ2の振幅設定で。理想的な断片サイズサイクル数は、第さまざまなサイクル数(例えば2、4、8、12、16、20サイクル以上)を試すことによって決定することができる。超音波処理装置の別のブランドは、しかし、条件は異なります使用してもよい。サイクル時間と量のオンとオフの数は実験が理想的な条件を決定するために行わなければならない。
- 超音波処理の結果を確認すると、定量化を行うために、各サンプルの20μlのを収集します。試料の残りを-80℃で保存することができる
- 0.1X TEバッファー30μlのを追加することによって、試料の20μlの希釈する。
- RNaseを1μlの試料を扱う37℃で1時間℃で、その後2時間62℃でプロテイナーゼKとインキュベートを1μlを追加します。
- 2%アガロースゲルでサンプルを20μlを実行します。
- 次に光度計、分光光度計を用いて定量化するQIAクイックPCR精製キットを用いて試料の残量を精製する。
*ネイティブCHIについてP、microccocalヌクレアーゼ消化を剪断するDNAを用いてもよい。
4。ブロックアガロースビーズ*
- ビーズがすでにブロックされている場合、5.1に進みます。
- プロテインAまたはプロテインGアガロースを使用してください。 5免疫沈降(IPアドレス)の場合、50%ビーズスラリー(300μlのビーズペレット)600μlのを使用
- ビーズを洗浄するために、4℃で1分間800回転でそれらをスピンダウンし、上清を捨てる。ゆっくり反転チューブが10倍で2ミリリットルチップ希釈バッファー(0.01%SDS、1.2mMのEDTA、167 mMのNaClを、1.1%トリトンX-100、16.7 mMのトリス-HCl pHは8.1)とのミックスよりも少し追加します。 4℃で1分間800回転で再びスピンダウンし、上清を捨てる。洗濯2回以上繰り返します。
- ブロッキング溶液に4℃で一晩回転させることによって、ビーズをブロックします。ブロッキング溶液のレシピについては、表1を参照してください。
5。プリ明確なクロマチン
- 雪解けは、氷の上でクロマチンを超音波処理した。
- 12,000 rpmでfでスピンダウンまたは4で10分間°C次にSDS(ホワイトペレット)を除去するためにすぐに氷の上に置く。
- 上清を収集し、ペレットを廃棄し、必要に応じてサンプルを結合。
- 計算(IPあたりのクロマチンの1-10 UG)に基づいて、実験に必要な量を取り出します。
- プロテイナーゼ阻害剤で補充ChIPの希釈バッファーで10倍クロマチンを希釈。
- IPごとブロックされるビーズを100μlを加える。
- 1時間4℃で回転させます。
6。免疫沈降
- 1分間800回転でサンプルをスピンダウンし、上清を新しいチューブに移す。
- 1分と別のきれいなチューブへの転送のために800rpmで上清をスピンダウン。
- -20℃で、入力制御として機能するように、上清20μlのを保存
- 実験で行われるIPアドレスの数にアリコートクロマチンを。
- 各サンプルにクロマチンの1-10 UGあたりの抗体の2 UGを追加します。
- 回転に一晩4℃でインキュベートする。
- 各IPサンプルにブロックされてビーズを100μlを加える。
- °C回転に4℃で1時間インキュベートする。
- ペレット1分間800 rpmでスピンダウンし、できるだけ上清の限りを捨てることによってビーズ。
- 低塩免疫複合体の洗浄緩衝液で一回ビーズを洗浄。各チューブに緩衝液1mlを加え、5-8分間室温で回転、1分間800 rpmでスピンダウンし、上清を捨てる。 5回洗浄した( 表2)の合計のためTE緩衝液で2回高塩免疫複合体の洗浄バッファーとLiClを免疫複合体の洗浄緩衝液で1回洗浄して繰り返します。
7。溶出
- 前日から解凍入力サンプルは、溶離液で処理することができます。
- 溶出バッファー新鮮( 表3)を作成します。
- IPと入力制御サンプルプラス1-2余分なサンプルのために必要な十分な溶出バッファーのマスターミックスを作る。
- 各IPサンプルに100μlの溶出バッファーを追加し、15メートルのために室温でインキュベート回転にした。
- 1分間800 rpmでスピンダウンし、新しいチューブに上清を追加します。
- ビーズの各チューブに溶出バッファーの別の100μLを加え、回転で15分間室温でインキュベートする。
- インキュベーション後15秒間ボルテックスし、1分間5,000 rpmでスピンダウンし、その後最初の溶出からの上清を上清兼ね備えています。 (不明な場合は、1分間5,000 rpmで再び上清をスピンダウン。ない上清中のビーズ上の残っていることを確認し、新しいチューブに上清を回収。
- 入力制御サンプルの20μlに溶出バッファー180μLを加える。
8。クロスリンクを逆転
- 溶離液と入力コントロールの200μlに、5 M NaClを8μLを加える。
- 65℃で一晩水浴中でパラフィルムとインキュベートとシールチューブ。
9。 DNA精製
- 37℃で1時間のRNase Aを1μlを各サンプルを扱う℃に
- 4μを追加0.5 M EDTA、8μlの1Mトリス-HCl、ミックス、lのはその後2時間45℃で各サンプルとインキュベートするプロテイナーゼKを1μlを追加します。
- QIAクイックPCR精製キットを用いて試料を精製する。サンプルは-20℃で保存することができ℃、PCR検査は後日に行うことができます。
*あるいは、ChIPのグレードの磁気ビーズ免疫沈降部分にアガロースの代わりに使用することができる。
10。 PCR検査
- PCR検査のために、目的のタンパク質によって結合することが知られている領域のためのプライマーを使用する。また、ネガティブコントロールとして非結合領域のためのプライマーを使用しています。
- 反応のための試薬を混ぜる。 1:100( 表3)で入力サンプルを希釈する。
- 反応を実行します。 PCRプログラム:
手順1:94℃3分
ステップ2:94℃20秒
59℃で30秒
72℃30秒
(少なくとも30 CYCLためのステップ2を繰り返します。ES)
ステップ3:72℃2分
- 1%アガロースゲルでサンプルを実行する。
- 濃縮はまた、リアルタイムPCRにより定量的に決定することができる。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
クロマチンは、超音波処理されており、RNアーゼおよびプロテイナーゼで処理された後、2%アガロースゲル上で実行するサンプルは、所望の大きさのDNAの大部分でスメアを提示しなければならない。いくつかの異なるサイクルが試験される場合、サイズが徐々に減少がサイクル数が増加する( 図2)として見られるべきである。
プロトコルの免疫沈降部が完了した後濃縮はどちらPCRまたはリアルタイムPCRにより確認することができる。アガロースゲル上で実行PCRサンプルでは(この場合はTCF7L2で目的のタンパク質に対する抗体を用いて、)入力とチップせいぜいサンプルレーンとは何か、に非常にかすかなバンド(バックグラウンドノイズ)にバンドがあるはずIgG抗体(陰性)陽性の結合領域の制御車線。負の結合領域のためのIgGコントロールとチップレーンのために非常にかすかな、あるいは全くバンドがあるはずです。入力レーン( 図3)にバンドがあるはずです。
図4は、リアルタイムPCRで調べた同じ試料を示す。前の図と同様に、IgGのコントロール上のChIPサンプルの陽性結合領域の著しい倍濃縮があるはずです。また、負の結合領域で見られるほとんどの濃縮、もしあれば、そこにあるべきである。

図1。ChIPのプロセスのフロー図は大きい数字を表示するには、ここをクリックしてください 。
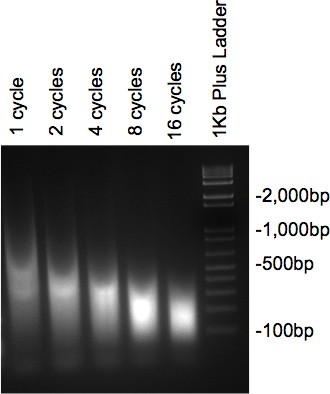
図2。ゲルDNAの超音波処理を確認してください。

図3。 PCRは、チップのチェック。
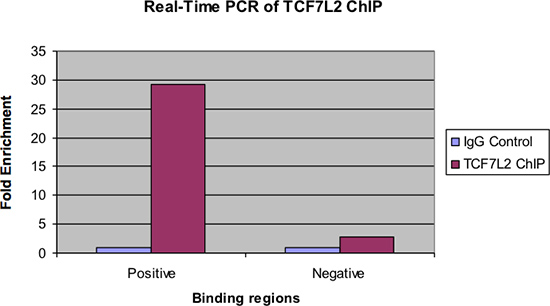
図4。 TCF7L2チップのリアルタイムPCR。
| 試薬 | ボリューム |
| ビーズペレット | 300μlの |
| BSA(50 mg / ml)で | 30μlの |
| 100Xプロテイナーゼ阻害剤 | 10μlの |
| ChIPの希釈バッファー | 660μlの |
| 合計 | 1,000μL |
表1。アガロースを遮断するためのレシピ。
| バッファ | コンポーネント |
| 低塩免疫複合体洗浄バッファー | 0.1%SDS 1%トリトンX-100 2mMのEDTA 20mMのトリス-HCl pHが8.1 150mMのNaCl |
| 高塩免疫複合体洗浄バッファー | 0.1%SDS 1%トリトンX-100 2mMのEDTA 20mMのトリス-HCl pHが8.1 500mMのNaClを |
| LiClを免疫複合体洗浄バッファー | 0.25 MのLiCl 1%NP-40 1%デオキシコール酸 1mMのEDTA 10mMトリス - 塩酸pH8.1の |
| TEバッファー | 10mMトリス - 塩酸pH8.1の 1mMのEDTA pH8.0で |
表2。 ChIPの洗浄バッファー。
| 試薬 | ボリューム |
| 10μlの | |
| 1 MのNaHCO 3 | 20μlの |
| H 2 O | 170μlの |
表3。つのIPのための溶出バッファー。
| 試薬 | 50μlの反応 | 20μlの反応 |
| 水 | 27μlの | 10.8μlの |
| 5X PCR反応緩衝液 | 10μlの | 4μlの |
| MgCl 2を | 4μlの | 1.6μL |
| のdNTP(10mM)を | 1μlの | 0.4μL |
| プライマーミックス(5μMの各) | 2μlの | 0.8μL |
| Taqポリメラーゼ(プロメガHotStartの) | 1μlの</ TD> | 0.4μL |
| のChIP DNA | 5μlの | 2μlの |
表4。 PCRの反応容量。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
ごく最近他の転写因子2,3で実証されているように、それは、現在のChIP-以降を用いてタンパク質-DNA相互作用アソシエーションのゲノムワイドなプロファイルを行うことが可能である。成功したシークエンシングの結果への鍵は、高品質のクロマチン免疫沈降DNAテンプレートの生成である。
DNAテンプレートが生成され、適切に濃縮されたことが確認された後、一方は、その後の配列決定のためのライブラリ調製にそれを取ることができる。例えば、一つは、ベンダー、イルミナが提供するシーケンシングライブラリプロトコルを使用することができます。このライブラリのサイズの選択は〜200のゲル電気泳動およびDNAのその後の切除および精製を行うことができる - 700-bpの範囲に。サイズを縮小し、ゲル精製から収集DNAのサイズ範囲は、チップの位置、seqの分解能を改善することを意図している狭める。 int型の要因にバインドされた入力DNAの小さな断片のために豊かにすることにより、erest、人は、そのサイトの場所は解像度を得ることが期待される。タイトなサイズ選択はまた、イルミナプラットフォームで生産分子コロニーの大きさの均一性を向上させます。このようなコロニーの大きさの均一性も得られる効果的な読み取り数が増加します。短いインプットDNAのサイズもイルミナプラットフォーム上でより堅牢なコロニーを生成し、これは任意の所与の入力サンプル分布内短いDNA片が同じ分布から長い入力個よりも最終的なシーケンス出力でより効率的に表現されることを意味する。
"次世代"配列解析へのバイオインフォマティクスのアプローチは、多くのベンダーは、さらなる改良のための彼らのソフトウェアのオープンソースを作ると、進化を続けています。一つは、DNA断片のオーバーラップのプロファイルにユニークなゲノムの場所にそのマップを読み込み、変換することができます。有意なピークはおおよそ偽発見率と同等の高さにthreshholdingプロファイルによって識別することができる。ポジションSPこの研究に由来するecific周波数マトリックスは、所定のファクタのヒトゲノムDNAを横切る結合部位を同定し、ローカライズするために使用することができる。
しかし、一つは、チップ配列と一緒に勉強するためにどのような要因が1希望に関して慎重でなければなりません。貧しい抗体が自分の実験結果に非常に有害な影響を持つことができるような抗体は、ChIPの設定で使用可能である市場で入手可能であればそのような研究に着手する前に、一方は評価すべきである。研究対象のタンパク質のスプライスアイソフォームが存在する場合に加えて、人は考えるべきである確かに、TCF7L2は、我々は、この転写因子のすべての主要なアイソフォームで一貫して存在するアミノ酸に結合した抗体を選択することで特に慎重であったので、多くのアイソフォームを有することが知られている25。
要約すると、クロマチン免疫沈降法および超ハイスループットシークエンシング(配列番号のChIP-)との組み合わせを識別することができ、所定の組織または、cでマップタンパク質-DNA相互作用エルライン。我々は、その後の配列決定のための高品質のChIPテンプレートを生成する方法を概説している。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者らは、彼らが競合する経済的利益を持っていないことを宣言します。
Acknowledgments
仕事はフィラデルフィアの小児病院から研究所開発賞によってサポートされています。
Materials
| Name | Company | Catalog Number | Comments |
| QIAquick PCR Purification Kit | Qiagen | 28104 | |
| EZ-ChIP Kit | Millipore | 17-371 | |
| GoTaq Hot Start Polymerase | Promega | M5001 | |
| Misonix Sonicator | Qsonica | XL-2000 | |
| NanoDrop 1000 Spectrophotometer | Thermo-Scientific | ||
| Positive control primer sequences (TCF7L2-1) Forward- 5'-TCGCCCTGTCAATAATCTCC-3' Reverse- 5'-GCTCACCTCCTGTATCTTCG-3' Negative control primer sequences (CTRL-1) Forward-5'-ATGTGGTGTGGCTGTGATGGGAAC-3' Reverse- 5'-CGAGCAATCGGTAAATAGGTCTGG-3' |
|||
References
- Odom, D. T., et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 303, 1378-1381 (2004).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316, 1497-1502 (2007).
- Robertson, G., et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature Methods. 4, 651-657 (2007).
- Reich, N. C., Liu, L.
- Lodige, I., et al. Nuclear export determines the cytokine sensitivity of STAT transcription factors. The Journal of Biological Chemistry. 280, 43087-43099 (2005).
- Schroder, K., Sweet, M. J., Hume, D. A. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology. 211, 511-524 (2006).
- Vinkemeier, U. Getting the message across, STAT! Design principles of a molecular signaling circuit. The Journal of Cell Biology. 167, 197-201 (2004).
- Brierley, M. M., Fish, E. N.
- Bentley, D. R.
- Grant, S. F., et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics. 38, 320-323 (2006).
- Sladek, R., et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 445, 881-885 (2007).
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447, 661-678 (2007).
- Saxena, R., et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 316, 1331-1336 (2007).
- Zeggini, E., et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 316, 1336-1341 (2007).
- Scott, L. J., et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 316, 1341-1345 (2007).
- Steinthorsdottir, V., et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nature Genetics. 39, 770-775 (2007).
- Salonen, J. T., et al. Type 2 Diabetes Whole-Genome Association Study in Four Populations: The DiaGen Consortium. American Journal of Human Genetics. 81, 338-345 (2007).
- Zeggini, E., McCarthy, M. I. TCF7L2: the biggest story in diabetes genetics since HLA. Diabetologia. 50, 1-4 (2007).
- Weedon, M. N.
- Hattersley, A. T. Prime suspect: the TCF7L2 gene and type 2 diabetes risk. The Journal of Clinical Investigation. 117, 2077-2079 (2007).
- Yochum, G. S., et al. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 104, 3324-3329 (2007).
- Duval, A., Busson-Leconiat, M., Berger, R., Hamelin, R. Assignment of the TCF-4 gene (TCF7L2) to human chromosome band 10q25.3. Cytogenet. Cell Genet. 88, 264-265 (2000).
- Pomerantz, M. M., et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nature Genetics. 41, 882-884 (2009).
- Tuupanen, S., et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nature Genetics. 41, 885-890 (2009).
- Zhao, J., Schug, J., Li, M., Kaestner, K. H., Grant, S. F. Disease-associated loci are significantly over-represented among genes bound by transcription factor 7-like 2 (TCF7L2) in vivo. Diabetologia. 53, 2340-2346 (2010).
- Benjamini, Y., Yekutieli, D. Quantitative trait Loci analysis using the false discovery rate. Genetics. 171, 783-790 (2005).
