

Summary
Retningslinjer for datamaskin basert strukturell og funksjonell karakterisering av protein bruke I-TASSER rørledning er beskrevet. Fra spørringen protein sekvens, er 3D-modeller genereres ved hjelp av flere tråder alignments og iterativ strukturell montering simuleringer. Funksjonelle slutninger er deretter trukket basert på kamper til proteiner med kjent struktur og funksjoner.
Abstract
Genomsekvensering prosjekter har kryptert millioner av protein sekvens, som krever kunnskap om deres struktur og funksjon for å bedre forståelsen av deres biologiske rolle. Selv om eksperimentelle metoder kan gi detaljert informasjon for en liten brøkdel av disse proteinene, er beregningsorientert modellering nødvendig for flertallet av protein molekyler som er eksperimentelt uncharacterized. I-TASSER server er et on-line arbeidsbenk for høy oppløsning modellering av protein struktur og funksjon. Gitt en protein sekvens, inkluderer en typisk output fra I-TASSER server sekundær struktur prediksjon, spådde løsemiddel tilgjengelighet av hver rester, homologe mal proteiner oppdaget av tråder og struktur justeringer, opptil fem helaftens høyere strukturelle modeller og struktur-basert funksjonell kommentarer for enzym klassifisering, Gene Ontologi vilkår og protein-ligand bindingssteder. Alle spådommene er merket med en tillit poengsum somforteller hvordan presise spådommer er uten å vite det eksperimentelle data. For å lette den spesielle forespørsler fra sluttbrukere, gir serveren kanalene til å akseptere brukerspesifiserte inter-rester avstand og kontakt kart til interaktivt endre I-TASSER modellering, det også tillater brukere å spesifisere noen proteiner som mal, eller å utelukke noen mal proteiner i løpet av strukturen montering simuleringer. Den strukturelle informasjonen kan samles inn av brukere basert på eksperimentelle bevis eller biologisk innsikt med den hensikt å forbedre kvaliteten på I-TASSER spådommer. Serveren ble vurdert som de beste programmene for protein struktur og funksjon spådommer de siste community-wide Casp eksperimenter. Det er for tiden> 20000 registrerte forskere fra over 100 land som bruker on-line I-TASSER server.
Protocol
Metode oversikt
Etter sekvensen til struktur-til-funksjon paradigme, innebærer jeg-TASSER prosedyre 1-4 for struktur og funksjon modellering fire påfølgende trinn: (a) mal identifikasjon av LOMETS 5, (b) fragment struktur remontering av replika- utveksle Monte Carlo simuleringer 6; (c) atomnivå struktur raffinement bruker REMO 7 og FG-MD-8, og (d) struktur-basert funksjon tolkninger bruke kofaktor 9.
Mal identifikasjon: For en spørring sekvens sendt inn av brukeren, er sekvensen første tres gjennom en representant PDB struktur biblioteket ved en lokalt installert LOMETS meta-threading server. Threading er en sekvens-struktur justering prosedyre som brukes for å identifisere mal proteiner som kan ha lignende struktur eller inneholder lignende strukturelle motiv som spørringen protein. For å øke dekningen av homologe templspiste påvisninger kombinerer LOMETS flere state-of-the-art algoritmer som dekker ulike threading metoder. Siden forskjellige threading programmene har forskjellige scoring systemer og justering sensitiviteter, er kvaliteten på den genererte threading alignments fra hver threading program vurderes av normalisert Z-score, som er definert som: 
hvor Z-score er scoren i standardavvik enheter i forhold til den statistiske gjennomsnittet av alle justeringer som genereres av programmet, og Z 0 er en program-spesifikke Z-score cutoff fastsettes basert på storstilt threading ytelsestester 5 til skille "god "og" dårlig "maler. En mal med en høy Z-score betyr at toppen malene har en innretting skårer signifikant høyere enn de fleste andre maler, som vanligvis innebærer at justeringen tilsvarer en god modell. Hvis de fleste av de beste threading malene har hiGH normalisert Z-score, er nøyaktigheten av den endelige I-TASSER modell vanligvis høy. Men hvis proteinet er stort og dekning av threading veielementer er begrenset til et lite område av spørringen protein, gjør en høy normalisert Z-score ikke nødvendigvis bety en høy modellering nøyaktighet for full-lengde modell. To øverste threading alignments fra hver threading program samles og brukes for neste trinn av struktur montering.
Iterativ struktur montering simulering: Etter threading prosedyren, er søket sekvensen delt inn threading justert og unaligned regioner. Kontinuerlig fragmenter i threading innretting er skåret fra maler og brukes direkte for strukturen montering, mens unaligned løkken regionene er bygd av ab initio modellering. Strukturen forsamlingen prosedyren er utført på et gitter system styrt av den kopi utveksling Monte Carlo simuleringer 6. I-TASSER kraftfelt inkluderer hydrogen-Bonding interaksjoner 10, kunnskapsbaserte statistisk energi vilkår avledet fra kjente proteinstrukturer i de 11 PDB, sekvens-basert kontakt spådommer fra SVMSEQ 12, og romlige begrensninger samlet fra LOMETS 5 threading maler. Den konformasjonsendring lokkeduer genereres i lav temperatur kopier i løpet av simuleringene er samlet av SPICKER 13 til å identifisere strukturer av lav fri energi stater. Cluster centroids av de beste klyngene er innhentet av gjennomsnitt 3D-koordinatene til alle gruppert strukturelle lokkeduer og brukes for den endelige modellen generasjon. Simuleringen og clustering prosedyren er gjentatt to ganger for å fjerne steric sammenstøt og videreforedling den globale topologi.
Atom-nivå modell konstruksjon og raffinement: Klyngen centroids oppnås etter SPICKER clustering er redusert protein modeller (hver rest representert ved sin C α og side-chain sentrum av masse) og have begrenset biologiske program. Byggingen av full-atom modell fra den reduserte modellene er gjort i to trinn. I det første trinnet, er REMO 7 brukes til å konstruere full atomære modeller fra C-alfa spor ved å optimalisere H-bindingen nettverk. I det andre trinnet, er REMO full-atomære modeller videreutviklet med 14 FG-MD, som forbedrer selve ryggraden torsjon vinkler, bindingslengdene, og side-chain rotamer orientering, med molekylære dynamiske simuleringer, som veiledet av den strukturelle fragmenter søkte fra PDB strukturer av TM-align. Den FG-MD raffinert modellene blir brukt som den endelige modeller for tertiær struktur spådommer ved I-TASSER.
Kvaliteten på den genererte modellene er estimert basert på en tillit score (C-score), som er definert basert på Z-score på LOMETS tråder justeringer og konvergens av I-TASSER simuleringer, matematisk formulert som: 
der
Den C-skår har en sterk sammenheng med kvaliteten på I-TASSER modeller. Ved å kombinere C-score og protein lengde, kan nøyaktigheten av de første I-TASSER modellene estimeres med en gjennomsnittlig feil på 0,08 for TM-score og 2 A for RMSD 15. Generelt, modeller med C-score> - er 1.5 forventes å ha en riktig fold. Her RMSD og TM-score er begge velkjente tiltak av topologiske likhet mellom modell og opprinnelige struktur. TM-score verdifullees varierer i [0, 1], hvor en høyere score indikerer en bedre struktur matche 16,17. Men for lavere rangerte modeller (dvs. 2 nd -5 th modeller), er korrelasjonen av C-score med TM-score og RMSD mye svakere (~ 0,5), og kan ikke benyttes for pålitelig estimering av absolutt modell kvalitet.
Er første modellen alltid den beste modellen i I-TASSER simuleringer? Svaret på dette spørsmålet avhenger av målet type. For enkle mål, er den første modellen som regel den beste modellen og dens C-score er vanligvis mye høyere enn resten av modellene. Men for harde mål, hvor threading ikke har sterk mal hits, er den første modellen ikke nødvendigvis den beste modellen, og jeg-TASSER faktisk har problemer med å velge den beste mal og modeller. Det anbefales derfor å analysere alle de 5 modellene for harde mål og velge dem basert på eksperimentelle informasjon og biologisk kunnskap.
Funksjon predictions: I det siste trinnet, er endelige 3D-modeller generert fra FG-MD brukes til å forutsi tre aspekter av protein funksjon, nemlig: a) Enzyme Commission (EC) nummer 18 og (b) Gene Ontologi (GO) 19 vilkår og ( c) bindingssteder for lite molekyl ligander. For alle de tre aspektene, er funksjonelle tolkninger generert ved hjelp av kofaktor, som er en ny tilnærming for å forutsi protein funksjon basert på globale og lokale likhet med mal proteiner i PDB med kjent struktur og funksjoner. Først er den globale topologien av forventet modeller matchet mot funksjonelle malbibliotekene bruke strukturelle justering program TM-align 20. Deretter blir et sett av proteiner mest lik målet modellene valgt fra biblioteket basert på deres globale struktur likheten, og et omfattende lokalt søk er utført for å identifisere struktur og sekvens likheten nær det aktive / binding nettstedet regionen. Den resulterende globale og lokale likheten score brukes til å rangeremal proteiner (funksjonell homologer) og overføre den merknaden (EC tall og Gene Ontologi 19 vilkårene) basert på toppen scoring treff. Tilsvarende er ligand binding stedet rester og ligand binding modus utledes basert på lokale tilpasning av spørring med kjent ligand binding nettstedet rester i toppen scoring funksjon maler 9.
Kvaliteten av funksjon (EF og GO term) prediksjon i I-TASSER er evaluert basert på funksjonelle homologi score (Fh-score) som er et mål på global og lokal likhet mellom søket og mal, og er definert som: 
der C-score er et estimat av kvaliteten på spådd modell som definert i Eq. (2), TM-score måler globale strukturelle likheten mellom modell og mal proteiner; RMSD ali er RMSD mellom modellen og malen strukturen i strukturelt innrettet regionen fra TM-align 20; Cov representerer dekning av strukturell tilpasning (dvs. forholdet mellom strukturelt justert rester dividert med spørringen lengde); ID ali er sekvensen identitet i TM-align justering. Den estimerte tillit score for EC nummer spådommer inneholder også et begrep for å vurdere aktive området kamp (ACM) mellom spørring og mal innenfor et definert lokalt område, beregnet som: 
der N t representerer antall mal rester til stede i det lokale området, N Ali nummeret til justert spørringen-template rester par, er d ii C α avstanden mellom i th par av sammenstilte rester, er d 0 = 3.0 Å avstanden cutoff, er M ii den BLOSUM score mellom ITH par av sammenstilte rester. Generelt er Fh-score i området [0, 5] og ACM score er mellom [0, 2], Hvor høyere score indikerer mer selvsikker funksjonell oppdrag. ACM poengsum er også brukt for å vurdere lokale struktur og sekvens likheten nær ligand-bindende områder, som er referert til som BS-score.
1. Innlevering av protein sekvens
- Besøk I-TASSER nettsiden på http://zhanglab.ccmb.med.umich.edu/I-TASSER å begynne med struktur og funksjon modellering eksperiment.
- Kopier og lim inn aminosyre sekvens i den medfølgende skjemaet eller direkte laste det opp fra din datamaskin ved å klikke på "Browse"-knappen. I-TASSER server godtar tiden sekvenser med opp til 1500 rester. Proteiner lengre enn 1500 rester er vanligvis multi-domene proteiner, og anbefales å deles opp i enkelte domener før innsending til I-TASSER.
- Gi din e-postadresse (obligatorisk) og et navn for jobben (valgfritt).
- Brukere kan eventuelt spesifisere eksterne inter-residue kontakt / avstand begrensninger, add-in en ekstra mal eller ekskludere noen mal proteiner i løpet av strukturen modellering prosessen. Lær mer om å bruke disse alternativene i "Discussion"-delen.
- For å sende sekvensen, klikker du på "Run I-TASSER"-knappen. Nettleseren vil bli sendt til en bekreftelse side som viser brukeren spesifisert informasjon, jobb identifikasjon (Job ID) nummer og en lenke til en nettside hvor resultatene vil bli deponert etter fullført jobb. Brukere kan bokmerke denne linken eller notere jobben identifikasjonsnummer for fremtidig referanse.
2. Tilgjengelighet av resultater
- Sjekk statusen for sendt jobb ved å gå til I-TASSER kø siden på http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Klikk på Søk-fanen og bruke Job ID-nummer eller spørringen sekvensen til søket sendt jobb.
- Etter struktur og funksjon modeling er ferdig, vil en e-post med bilde av spådd strukturer og en web-link bli sendt til deg. Klikk på denne linken eller åpne koblingen bokmerkede på trinn 1,5 til vise og laste ned resultatene.
3. Sekundær struktur og løsemiddel tilgjengelighet spådommer
- Sjekk FASTA formaterte spørringen sekvens vises på toppen av resultatet siden. Hvis noen ekstra tilbakeholdenhet / malen ble spesifisert under sekvens underkastelse, en link til nettsiden som viser brukeren spesifisert informasjon kan også sees (Figur 1A).
- Undersøk sekundære strukturen prediksjon vises som: alfa heliks (H), beta Strand (S) eller spole (C) og tillit score på prediksjon (0 = lav, 9 = høy) for hver rester. Se etter region med lange strekninger av vanlig sekundær struktur (H eller S) spådommer, til å anslå kjerne-regionen i protein. Strukturelle klasse av protein kan også bli analysert basert på fordelingen av sekundære strukturer elementer. Alså lenge regioner av coil elementer i protein vanligvis indikerer ustrukturerte / uordnede regioner.
- Se den predikerte solvent tilgjengelighet (figur 1C) å fastslå begravd og væske utsatte regioner i søket. Verdier av forventet løsemiddel tilgjengelighet varierer fra 0 (nedgravde rester) til 9 (utsatt rest). Region inneholder stort sett begravd rester kan brukes til å avgrense kjernen regionen i protein, mens regioner med løsemiddel eksponert og hydrofile rester er potensielle hydrering / funksjonelle områder.
Fire. Tertiær struktur spådommer
- Scroll ned for å vise spådd tertiær strukturer av spørringen protein, vises i en interaktiv Jmol applet (figur 2). Venstre klikk på appleten for å endre utseendet til viste struktur, zoom inn bestemt region, velge bestemte rester typer i den predikerte modell eller beregne inter-rester avstander.
- Analyser modeller for tilstedeværelsen av lange ustrukturert regioner. Disse regions vanligvis tilsvarer disordered regioner i protein eller indikerer mangel på mal justering. Disse regionene har generelt lav modellering nøyaktighet og fjerne disse regionene under modellering av N & C-terminus regionen vil forbedre modellering nøyaktighet.
- Last ned PDB formaterte strukturen filer av modellen ved å klikke på "Download Model" linker. Du kan åpne disse filene i en molekylær visualisering programvare (f.eks Pymol, Rasmol etc.) for videre analyse av strukturelle trekk.
- Analyser tillit score (C-score) av strukturen modellering for å anslå kvaliteten på spådd strukturer. C-score (Eq. 2) verdier er typisk i området [-5, 2], hvor en høyere poengsum avspeiler en modell av bedre kvalitet. Den estimerte TM-score og RMSD av første modellen er vist som "Beregnet nøyaktigheten av Model 1". For lange proteiner, er det anbefalt å evaluere modellen kvalitet basert på TM-score, som TM-score er mer følsom for den topologiske endringer enn RMSD. < li> Klikk på "mer om C-score" linken for å analysere C-score, cluster størrelse og cluster tetthet av alle modellene. Beregnet TM-score og RMSD presenteres kun for det første I-TASSER modell, fordi C-score på lavere rangert modeller ikke er sterkt korrelert med TM-score eller RMSD. Kvaliteten på lavere rangert modellene kan delvis vurderes basert på deres cluster tetthet og cluster størrelse i forhold til den første modellen, der modeller fra større cluster og høyere tetthet er i gjennomsnitt nærmere den opprinnelige strukturen.
- Low C-score spådommer vanligvis indikerer en lav nøyaktighet prediksjon. I de fleste slike tilfeller mangler spørringen protein en god mal i biblioteket, og har en størrelse utenfor rekkevidden av ab initio modellering (dvs.> 120 rester). I disse tilfellene kan brukerne søke for ekstra romlige begrensninger og bruke dem til å forbedre I-TASSER modellering (se Discussion avsnitt). Det er også oppfordret til å sende sekvenser til vår QUARK server (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) for en ren ab initio modellering hvis proteinet størrelsen er under 200 rester.
5. LOMETS target mal innretting
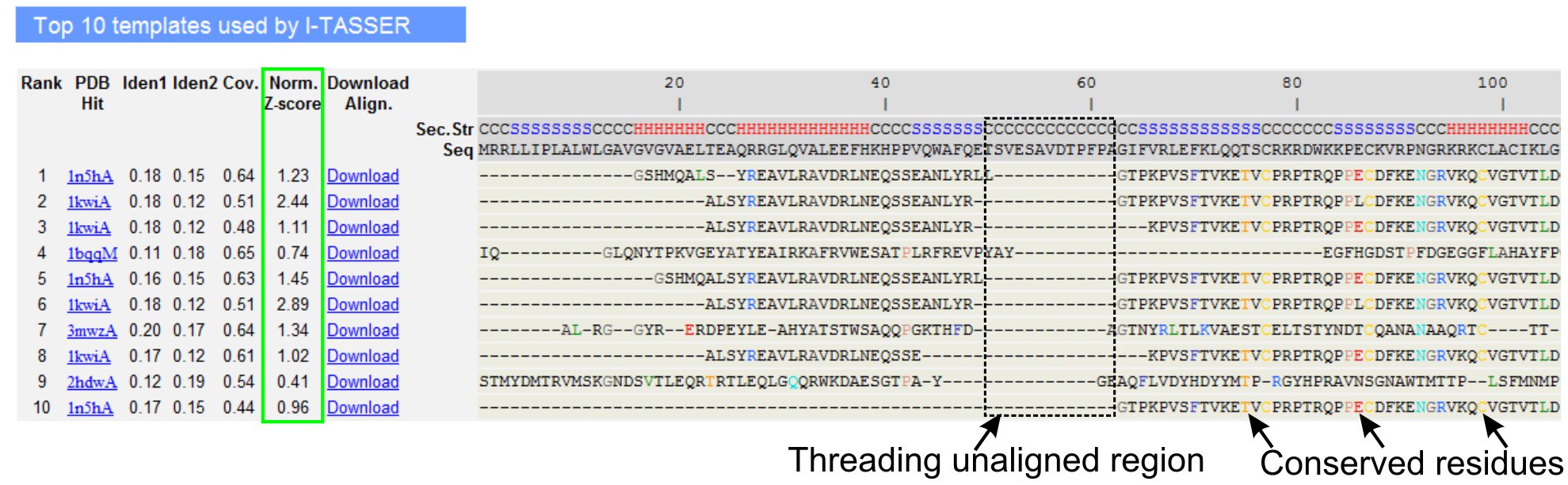
- Scroll ned for å analysere de ti beste threading maler av spørringen protein, som er identifisert av LOMETS threading programmer (figur 3). Vis normaliserte Z-score (Eq. 1), vist i "Norm. Z-score "kolonnen til å analysere kvaliteten på threading justeringer. Justeringer med en normalisert Z-score> 1 gjenspeiler en trygg innretting og mest sannsynlig ha samme fold som spørringen protein.
- Analyser sekvensen identitet i threading alliansefrie regionen (kolonne 'Iden. 1') og for hele kjeden (kolonne 'Iden. 2') for å vurdere homologi mellom søket og malen proteiner. Høy sekvens identitet er en indikator på evolusjonært slektskap mellom søket og mal proteiner.
- Vis threading justert rester vist i farget for å visuelt identifisere ulempererved rester / motiver i søket og malen proteiner. En høyere sekvens identitet i threading-justert regionen, i forhold til hel-kjeden justering indikerer også tilstedeværelsen av bevart strukturelle motiv / domener i spørringen.
- Vurdere dekning av threading justering ved å se på "Cov. kolonne og inspisere justeringen. Dersom dekningen av topp justeringer er lav og begrenset til bare en liten region av spørringen protein eller fraværende for en lengre segment av spørringen sekvens, deretter spørringen protein inneholder som regel mer enn ett domene, og det anbefales å splitte sekvensen og modell domenene individuelt (figur 3).
- Last ned PDB formaterte sekvens-struktur justering filer ved å klikke på "Download Align" koblinger. Disse innretting Filen kan åpnes i alle molekylær visualisering program oppført i Materials delen, og kan også brukes for å legge til flere begrensninger i strukturen modellering (Step 1,4).
Seks.Strukturelle analogs i PDB
- Vis den neste tabellen (figur 4) av resultatet siden for å bestemme de ti beste strukturelle analoger av de første spådd modellen, som identifiseres ved det strukturelle justering programmet TM-align 20. En TM-score> 0,5 indikerer at det oppdagede analog og modell har en lignende topologi og kan brukes til å bestemme strukturelle klassen / protein familie av spørringen protein 16, mens de med TM-score <0,3 betyr en tilfeldig struktur likhet.
- Analyser sekvensen identitet og RMSD i strukturelt justert regionen er vist i 'IDEN en' og 'RMSD en "kolonnene for å vurdere bevaring av romlige motiver i modellen og den strukturelle analog. Inspiser de fargede og justert rester par i justeringen for å identifisere disse strukturelt bevart rester og motiver.
- Klikk på PDB koden som vises i "PDB Hit"-kolonnen for å besøke RCSB nettsted og lære mer om deres strukturelle klassifisering (SCOP, Cath og PFAM) og funksjonell informasjon (EC nummer, tilknyttede GO vilkår og bundet ligand).
7. Funksjon prediksjon bruke kofaktor
- Bla nedover i resultatet siden å analysere funksjonelle tolkninger for spørringen protein. Protein funksjoner er oppregnet i tre sammenheng tabeller, viser: Enzyme Commission (EC) tall, Gene Ontologi (GO) termer, og ligand bindingssteder.
- Vis 'TM-score', 'RMSD a', 'IDEN en' og 'Cov.' kolonnene i hver tabell for å analysere parametere av global struktur likhet og bevaring av romlige mønstre mellom modell og identifisert funksjonelle homologer (templates).
8. Enzyme Commission nummer prediksjon
- Se de fem potensielle enzym homologer av spørringen protein vises i "Predicted EF tall" table (figur 5). Tilliten nivå av EC nummer prediksjon ved hjelp av disse malene er vist i "EC-Resultat-kolonnen. Basert på benchmarking analyse 23, funksjonelle likheten (første tre sifrene i EC nummer) mellom søket og mal protein kan bli pålitelig tolkes med EC-score> 1.1.
- Se etter konsensus av funksjon (EF tall) blant de maler, som har lignende fold (dvs. TM-score> 0,5) som spørringen protein. Hvis flere maler har samme EC nummer og EC-score> 1.1, er det konfidensnivå på prediksjon svært høy. Men hvis EC-Score er høy, men det er en mangel på konsensus blant de identifiserte hits, så prediksjon blir mindre pålitelig og brukere anbefales å konsultere GO sikt spådommer.
- Klikk på linken gitt på EC for å besøke ExPASy enzym databasen og analysere funksjon, inkludert reaksjonen katalysert, co-faktor krav og metabolismevei, av malen protein i detalj.
9. Gene Ontologi (GO) term spådommer
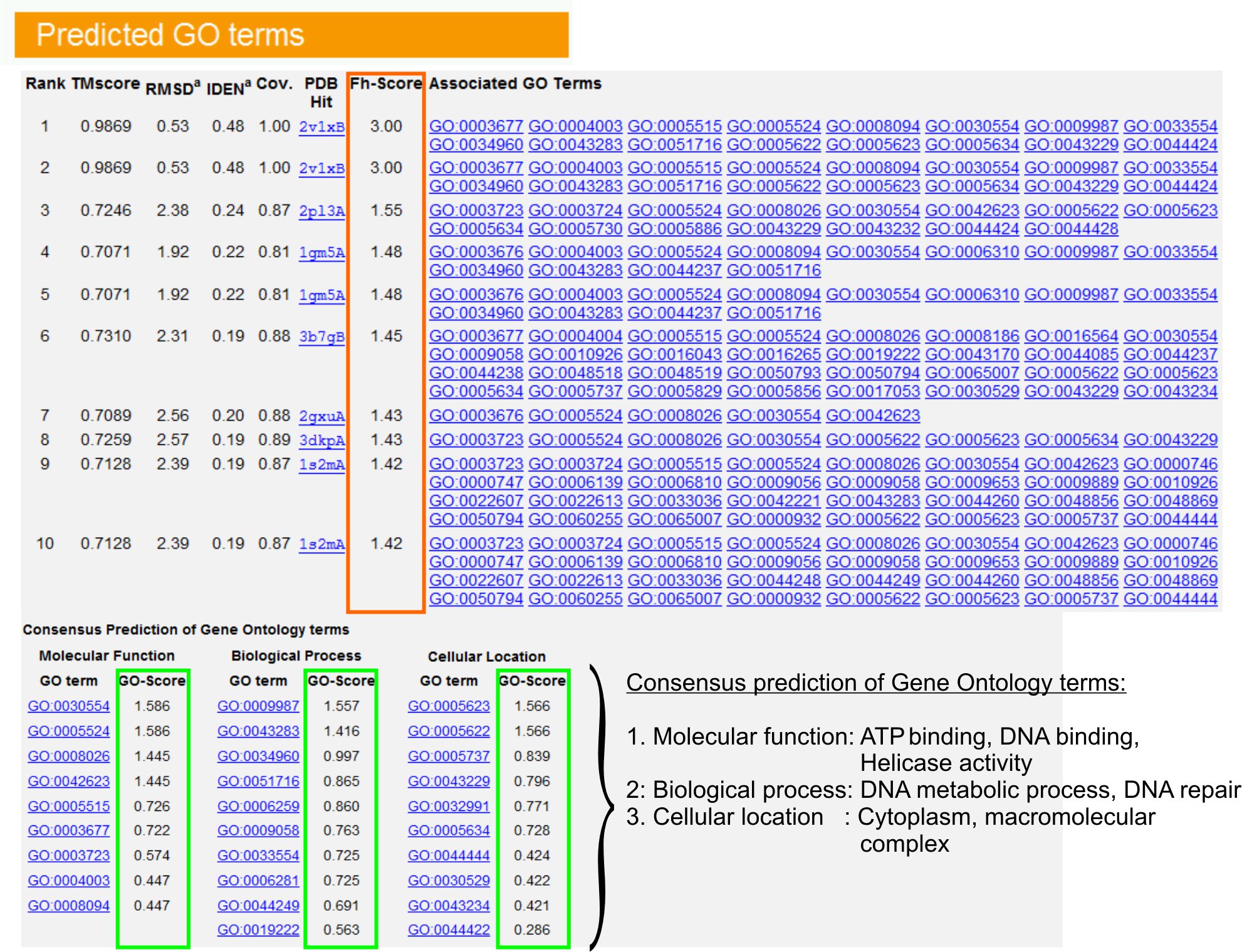
- Se de "Predicted GO begrepene" table (Figure 6) for å identifisere topp ti homologer av spørringen protein i PDB bibliotek, kommenterte med Gene Ontologi (GO) vilkår. Hvert protein er vanligvis assosiert med multiple GO vilkår, som beskriver sin molekylære funksjoner (MF), biologiske prosesser (BP) og cellulær komponent (CC). Klikk på hvert semester å besøke Amigo nettsiden og analysere sin definisjon og ætt.
- Analyser Fh-score (Functional homologi score) kolonnen for å få tilgang til funksjonelle likheten mellom søket og mal proteiner og beregne sikkerhetsnivå for overføring funksjonell annotering av disse proteinene. I vår benchmarking studie 23, kunne 50% av de innfødte GO vilkårene være korrekt identifisert fra første identifiserte malen ved hjelp av en Fh-score cutoff på 0,8, med en samlet nøyaktighet på 56%.
- Se den "Consensus prediksjon av GO Begrepene" bordet for å analysere sammenfall av funksjon mellom maler. Disse vanlige funksjoner brukes for å forutsi den GO betingelser (MF, BP og CC) av spørringenprotein og vurdere sikkerhetsnivå (GO-score) av GO sikt spådommer. Basert på benchmarking test 23, er de beste falske positive og falske negative priser innhentet for spådommer med GO-skår cutoff = 0,5, med synkende dekning av prediksjon på dypere ontologi nivåer.
10. Protein-ligand binding stedet spådommer
- Rull ned til bunnen av siden for å se topp ti ligand binding nettstedet spådommer for spørringen protein. Spådd bindingssteder rangeres basert på antall spådd ligand conformations som deler felles forpliktende lomme. Den beste identifiserte bindende området er allerede vist i Jmol applet. Klikk på radioen for å analysere andre spådommer og visualisere ligand samspill rester.
- Analyser BS-score kolonnen for å evaluere lokale likheten mellom modellen og malens bindende nettsted. Basert på referanseporteføljen 9, BS-score> 1,1 indikerer høy sekvens og struktur similarity nær spådd bindende området i modell og kjent bindende stedet i malen.
- Last ned PDB formatert strukturen fil av komplekset ved å klikke på "Download" linken. Brukere kan åpne disse filene i valgfri molekylær visualisering program og interaktivt vise den anslåtte bindende nettstedet og ligand-protein interaksjoner på sin lokale datamaskin.
11. Representative resultater

Figur 1 Et utdrag fra I-TASSER siden, som viser (A) FASTA formatert søket sekvens;. (B) spådde sekundær struktur og tilhørende selvtillit scorer, og (C) spådde løsemiddel tilgjengelighet av rester. Analysert kjerne regionen og potensielle hydrering stedet i søket er uthevet i cyan og røde rektangler, henholdsvis.

Figur 2.

Figur 3. Et eksempel på I-TASSER siden, som viser topp ti identifisert threading maler og linjer ved LOMETS 5 tråder programmer. Kvaliteten på threading veielementer er evaluert basert på normalisert Z-score (uthevet i grønt), hvor en verdi> 1 reflekterer en trygg justering. Justert rester i malen som er identiske med de tilsvarende spørringen rester er markert med farge for å indikere tilstedeværelse av bevarte rester / motiv, mens en mangel på justering i de fleste av topp maler indikerer tilstedeværelse av flere domener i søket protein og unaligned rester korresponderer til domenet linker regioner. Klikk her for å se full størrelse versjon av figur 3.

Figur 4. Et eksempel på siden, som viser topp ti identifisert strukturelle analoger og strukturelle justeringer, identifisert av TM-align 20 strukturell justering program. Rangeringen av analogs vist er basert på TM-score (uthevet i blått) av de strukturelle justering. En TM-score> 0,5 indikerer at de to sammenlignet strukturene har en lignende topologi, mens en TM-score <0,3 betyr en likhet mellom to tilfeldige strukturer. Strukturelt justert rester parene er uthevet i farger basert på deres aminosyre-eiendom, mens unaligned regionene er angitt med "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Klikk her for å se full størrelse versjon av figur 4.

Figur 5. Et eksempel på I-TASSER siden, som viser identifiserte enzym homologer av spørringen protein i PDB biblioteket. Tilliten nivå av EC nummer prediksjon er analysert basert på EC-score (uthevet i grønt), hvor EC-score> 1,1 indikerer funksjonelle likheten (samme første tre sifrene i EC nummer) mellom søket og mal protein.

Figur 6. Et eksempel på I-TASSER siden, som viser GO sikt spådommer for spørringen protein. Funksjonell homologer for søket protein i Gene Ontologi Malbibliotek rangeres basert på deres Fh-score (i oransje rektangel). Felles funksjonelle egenskaper fra disse topp-scoring hits er avledet til gene spiste den endelige GO sikt spådommer for spørringen protein. Kvaliteten på de predikerte GO vilkårene er estimert basert på GO-score (vist i grønt), hvor en GO-score> 0,5 indikerer en pålitelig prediksjon. Klikk her for å se full størrelse versjon av figur 6.

Figur 7. Et eksempel på I-TASSER siden, som viser de ti beste protein ligand binding nettstedet spådommer bruke kofaktor 9 algoritmen. Rangeringen av forventet bindingssteder er basert på antall spådd ligand conformations som deler felles forpliktende lomme i søket. BS-score (uthevet i rødt) er et mål på lokalt sekvens og struktur likheten mellom de predikerte og malens bindende område, og er nyttig for å analysere bevaring av bindende nettstedet lommer.
les/ftp_upload/3259/3259fig8.jpg "/>
Figur 8. Et eksempel på eksterne tilbakeholdenhet filer som brukes til å spesifisere rest-rester kontakt / avstand begrensninger.

Figur 9. Eksempel på tilbakeholdenhet filer som brukes for å angi en mal protein til I-TASSER server. Brukeren kan spesifisere søket-template justering enten i (A) FASTA format, eller (B) 3D format.

Figur 10. Et eksempel filen som brukes for å utelukke mal under I-TASSER struktur modellering prosedyre. Den første kolonnen inneholder PDB ID malen proteiner for å bli ekskludert. Den andre kolonnen brukes til å angi rekkefølgen identitet cutoff som vil bli brukt til andre tilsvarende maler i malen biblioteket.
Discussion
Protokollen presentert ovenfor er en generell retningslinje for struktur og funksjon modellering ved hjelp av I-TASSER server. Selv om denne automatiserte fremgangsmåten fungerer svært godt for de fleste proteiner, menneskelige inngrep ofte hjelpe til å forbedre modellering nøyaktighet, spesielt for de proteinene som mangler nære maler i PDB biblioteket. Brukere kan gripe inn under I-TASSER modellering på følgende måter: (a) splitting av multi-domene proteiner, (b) å gi eksterne begrensninger for å forbedre strukturen forsamlingen, og (c) fjerne maler under modellering.
Splitting multi-domene protein:
Mange lange proteinsekvenser ofte inneholder flere domener tethered av fleksible linker regioner, noe som gjør deres strukturoppklaring vanskelig å bruke både eksperimentelle og beregningsorientert teknikker. Likevel, som domener er uavhengig folding enheter og kan utføre forskjellige molekylære funksjon, det erønskelig å splitte lange multi-domene proteiner og modell hvert domene separat. Modeling domener individuelt vil ikke bare øke hastigheten på prediksjon prosessen, men også øker kvaliteten på spørringen-template justering, noe som resulterer i mer pålitelig struktur og funksjon spådommer.
Domain grenser i protein sekvenser kan forutsies ved hjelp av fritt tilgjengelig eksterne online programmer som NCBI 24 CDD, PFAM 25 eller InterProScan 26. Likeledes, hvis LOMETS threading justeringer er tilgjengelig for spørringen protein, kan domenenavn grenser være plassert ved visuelt identifisere lange strekninger av unaligned rester i toppen threading maler (se trinn 5.4). Disse unaligned regionene meste korresponderer til domenet linker regioner. Hvis multi-domene maler er allerede tilgjengelig i malen PDB bibliotek med alle spørringen domenene justert, så spørringen protein kan modelleres som full lengde.
Gi eksterne begrensninger
A. Angi kontakt / avstand begrensninger
Eksperimentelt preget inter-rester kontakter / avstander, for eksempel fra NMR ellercross-linking eksperimenter, kan spesifiseres ved å laste opp en tilbakeholdenhet fil. Et eksempel filen er vist i Figur 8, hvor Kolonne 1 angir hvilken type tvang, dvs. "DIST" eller "kontakt". For avstand tilbakeholdenhet (DIST), kolonne 2 og 4 inneholder rester posisjoner (i, j), kolonne 3 og 5 inneholder atom-typer i rester og kolonne 6 angir avstanden mellom de to angitte atomer. For kontakt begrensninger (kontakt), kolonne 2 og 3 inneholder stillingene (i, j) av rester som bør være i kontakt. Avstanden mellom sidekjeder sentrum av disse kontakte rester parene avgjøres basert på observert avstander i kjente strukturer i PDB. I-TASSER vil prøve å trekke disse atom parene nær angitt avstand i løpet av strukturen raffinement simuleringer.
B. Angi en proteinstruktur mal
LOMETS threading programmene bruker en representant PDB biblioteket for å finne plausible folder for spørringen Protein. Selv med en representant struktur bibliotek bidrar til å redusere tiden som kreves for å beregne sekvens-struktur alignments, er det mulig at en god mal protein er savnet i biblioteket eller mal kan ikke ha blitt identifisert av LOMETS tråder programmer, selv om det er til stede i biblioteket. I slike tilfeller bør brukeren spesifisere ønsket protein struktur som mal.
Hvis du vil angi proteinstruktur som en ekstra mal, kan brukerne enten laste opp en PDB formatert struktur fil eller spesifisere PDB IDen til en deponert protein struktur i PDB bibliotek. I-TASSER vil generere spørringen-template justering bruker mønstre program 23 og vil samle romlige begrensninger både fra den angitte brukeren mal og LOMETS maler for å veilede strukturen forsamlingen simulering. Fordi nøyaktigheten av LOMETS innskrenkninger er forskjellig for ulike mål, er vekten av LOMETS begrensninger sterkere i lett (homologe) TArgets enn i hard (non-homolog) mål, som har blitt systematisk innstilt i vår benchmark trening.
Brukere kan også angi sin egen spørring-mal alignments. Serveren godtar justering i to formater: den FASTA format (figur 9A) og 3D-format (figur 9B). Den FASTA formatet er standard og beskrevet på http://zhanglab. ccmb.med.umich.edu / FASTA / . 3D-formatet er lik standard PDB format ( http://www.wwpdb.org/documentation/format32/sect9.html ), men to ekstra kolonner avledet fra malene er lagt til ATOM poster (se figur 9B):
Kolonner 10-30: Atom (C-alfa only) og rester navn for spørringen sekvensen.
Kolonner 31-54: Koordinater for C-alfa atomer av spørringen kopiert fra tilsvarende atomene i malen.
Kolonner 55-59: Tilsvarende rester nummer i malen basert på innretting
Kolonner 60-64: Tilsvarende rester navn i malen
Utelukk maler proteiner
Proteiner er fleksible molekyler og kan vedta flere konformasjonsendring stater til å endre sin biologiske aktivitet. For eksempel har strukturer av mange protein kinaser og membran proteiner løst i både aktive og inaktive konformasjon. Også tilstedeværelse eller fravær av bundet ligand kan føre til store strukturelle bevegelser. Mens alle de conformational statene malen er like for threading programmer, er det ønskelig å modellere søket ved hjelp av maler i bare én bestemt tilstand. Et nytt alternativ på serveren tillater brukeren å utelukke mal proteiner i løpet av struktur modellering. Denne funksjonen vil også tillate brukeren å velge homologi nivået av maler som skal brukes for modellering. Brukere kan utelukke mal proteiner from I-TASSER biblioteket ved:
A. Angi en sekvens identitet cutoff
Brukere kan bruke dette alternativet for å utelukke homologe proteiner fra I-TASSER mal bibliotek. Den homologi er satt basert på sekvens identitet cutoff, dvs. antall identiske rester mellom søket og malen protein delt sekvensen lengden av spørringen sekvensen. For eksempel, hvis brukeren skriver i "70%" i den medfølgende skjemaet, alle maler proteiner som har en sekvens identitet> 70% til spørringen protein jeg-vil bli ekskludert fra I-TASSER mal bibliotek.
B. Exclude bestemt mal proteiner
Bestemt mal proteiner kan bli ekskludert fra I-TASSER mal biblioteket ved å laste opp en liste som inneholder PDB IDene til strukturer for å bli ekskludert. Et eksempel filen er vist i Figur 10. Som samme protein kan eksistere som flere oppføringer i PDB bibliotek, I-TASSER server vil som standard utelukke den angitte maler (i COLUMN1) så vel som alle andre maler fra biblioteket som har en identitet> 90% til den angitte maler. Brukere kan også angi en annen identitet cutoff, f.eks 70%, hvor alle maler med identitet> 70% til spesifisert mal Proteinene vil bli ekskludert.
Disclosures
Ingen interessekonflikter erklært.
Acknowledgments
Prosjektet støttes delvis av Alfred P. Sloan Foundation, NSF Career Award (DBI 1.027.394), og National Institute of General Medical Sciences (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H.
- Finn, R. D.
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}