

Summary
I-TASSER boru hattı kullanarak protein yapısal ve fonksiyonel karakterizasyonu tabanlı bir bilgisayar için yönergeler açıklanmıştır. 3D modelleri sorgu protein dizisi başlayarak, birden fazla iş parçacığı hizalanmalar kullanılarak oluşturulan yapısal montaj simülasyonları yinelemeli. Fonksiyonel çıkarımlar sonra bilinen yapı ve fonksiyonlara sahip proteinlere maçlarına göre çizilir.
Abstract
Genom sıralama projeleri kendi biyolojik rolü anlayış geliştirmek için yapı ve fonksiyon bilgisi gerektiren, milyonlarca protein dizisi şifrelenmiştir. Deneysel yöntemler bu proteinlerin küçük bir kısmı için ayrıntılı bilgi sağlayabilir rağmen, sayısal modelleme, deneysel uncharacterized protein moleküllerinin çoğunluğu için gereklidir. I-TASSER sunucu protein yapısı ve işlevi yüksek çözünürlüklü modelleme için bir on-line bir tezgah. I-TASSER sunucu tipik bir çıkış, bir protein dizisi göz önüne alındığında, ikincil yapı tahmini içeren her bir kalıntı solvent erişilebilirlik, homolog parçacığı ve yapısı hizalanmalar tarafından tespit şablon proteinler, beş tam uzunlukta üçüncül yapısal modeller ve yapı tabanlı tahmin enzim sınıflandırma fonksiyonel ek açıklamalar, Gen Ontoloji açısından ve protein-ligand bağlanma yerleri. Tüm tahminler bir güven puanı ile etiketlenmiş olanöngörüleri deneysel verileri bilmeden ne kadar doğru söyler. Son kullanıcıların özel istekler kolaylaştırmak için, sunucu, kullanıcı tarafından belirtilen inter-kalıntı mesafe kabul ve I-TASSER modelleme etkileşimli değiştirmek için haritalar iletişim kanalı sağlayan, aynı zamanda kullanıcıların şablon olarak herhangi bir protein belirtmek için izin verir, veya herhangi bir şablon dışlamak için sırasında proteinlerin yapı montaj simülasyonları. Yapısal bilgiler I-TASSER tahminlerin kalitesinin iyileştirilmesi amacı ile deneysel kanıtlar veya biyolojik anlayışlar dayalı kullanıcıları tarafından toplanmış olabilir. Sunucu toplum genelinde son CASP deneylerde protein yapısı için en iyi programlar ve fonksiyon tahminlerde olarak değerlendirildi. Şu anda on-line I-TASSER sunucu kullanıyorsanız, 100'den fazla ülkeden gelen 20.000 kayıtlı bilim adamları vardır.
Protocol
Yöntem genel bakış
Yapısı ve fonksiyonu modelleme için I-TASSER prosedürü 1-4 sıra-yapı-fonksiyon paradigma ardından arka arkaya dört adım içerir: (a) LOMETS 5 şablon tanımlama; (b) takılması ile çoğaltma parçası yapısı Döviz Monte Carlo simülasyonları 6; REMO 7 ve FG-MD 8 (c) atomik seviyede yapı arıtma; ve (d) kofaktör 9 kullanılarak yapı-fonksiyon yorumlara.
Şablon tanımlama: kullanıcı tarafından gönderilen bir sorgu sırası için sırası ilk meta-Threading sunucusu yerel olarak yüklü bir LOMETS tarafından bir temsilci PDB yapısı kütüphanesi ile dişli. Threading benzer bir yapıya sahip veya sorgu protein olarak benzer yapısal motif içerebilir şablon proteinleri tanımlamak için kullanılan bir dizi yapı hizalama işlemdir. Homolog Templ kapsama alanını artırmak içinyedik tespitler, LOMETS birden fazla iş parçacığı farklı metodolojiler kapsayan state-of-the-art algoritmalar birleştirir. Farklı parçacığı programları farklı puanlama sistemleri ve hizalama hassasiyetleri olduğundan, her iş parçacığı program oluşturulan iş parçacığı ittifaklarının kalite olarak tanımlanan normalleştirilmiş Z-skoru, tarafından değerlendirilir : 
Z-skoru program tarafından oluşturulan tüm hizalanmalar istatistiki ortalamaya göre standart sapma birimleri puan ve Z 0 'iyi ayırt etmek için büyük ölçekli iş parçacığı benchmark testleri 5 dayalı belirlenen bir programa özgü Z-skoru kesme 've' kötü 'şablonlar. Yüksek bir Z-skoru ile bir şablon üst şablonları genellikle hizalamayı iyi bir model karşılık geldiğini ima diğer şablonları, en anlamlı derecede daha yüksek bir hizalama puanı var demektir. Üst parçacığı şablonlar çoğu hi varsanormalize gh Z-skorları, son I-TASSER modelinin doğruluğunu genellikle yüksektir. Ancak, proteinin büyük ve kapsama parçacığı hizalanmalar sorgu protein küçük bir bölge ile sınırlı ise, yüksek bir normalize Z-skoru mutlaka tam uzunlukta model için yüksek bir modelleme doğruluğu anlamına gelmez. Her parçacığı programından en iyi iki iş parçacığı hizalanmalar yapı montaj sonraki adım için toplanır ve kullanılır.
Yapısı montaj simülasyon Iteratif parçacığı işlemi sonrasında, sorgu sırasını hizalanmış ve hizalanmamış bölgelerde parçacığı ayrılmıştır. Sürekli parçacığı hizalama parçaları hizalanmamış döngü bölgeler ab initio modelleme tarafından inşa ederken, şablonlar eksize ve yapı montajı için doğrudan kullanılır. Çoğaltma döviz Monte Carlo simülasyonları 6 tarafından yönlendirilen bir kafes sistemi yapısı montaj işlemi yapılır. I-TASSER kuvvet alanında hidrojen-boetkileşimleri 10, 11, PDB SVMSEQ 12 sıra-tabanlı iletişim öngörüleri ve LOMETS 5 parçacığı şablonları toplanan mekansal sınırlamalar bilinen protein yapılarının türetilmiş bilgi tabanlı istatistiksel enerji açısından bulgu. Simülasyonlar sırasında düşük sıcaklık kopyaları konformasyonel tuzakları düşük serbest enerji devletlerin yapılarını belirlemek için SPICKER 13 kümelenmiş. Kümelenmiş tüm yapısal tuzakları 3D koordinatları ortalama ve son model üretimi için kullanılan üst kümelerin Küme sentroidler elde edilir. Sterik çatışmalar kaldırılması ve küresel topoloji daha rafine simülasyon ve kümeleme işlemi iki kez tekrarlanır.
Atom seviyesi model inşaat ve arıtma: SPICKER kümeleme sonra elde edilen küme sentroidler protein modelleri (C α ve yan zinciri kütle merkezi tarafından temsil edilen her bir kalıntı) ve h azalırave sınırlı biyolojik uygulama. Azaltılmış modelleri tam atom modeli yapımı, iki adımda yapılır. İlk adımda, REMO 7 H-bağı ağları optimize ederek C-alfa izleri tam atom modelleri oluşturmak için kullanılır. İkinci adımda ise, REMO tam atom modelleri, omurga torsiyon açıları, bağ uzunlukları, ve yan zincir rotamer yönelimleri geliştirir FG-MD 14, rafine, moleküler dinamik simülasyonları tarafından aranır yapısal parçaları tarafından yönlendirilen tarafından PDB yapıları TM-align. FG-MD rafine modelleri I-TASSER üçüncül yapısı tahminlerin son model olarak kullanılmaktadır.
LOMETS parçacığı hizalanmalar Z-skoru ve yakınsama I-TASSER simülasyonları, matematiksel olarak formüle dayalı tanımlanan bir güven puanı (C puanı), dayalı oluşturulan modellerin kalitesi tahmin edilmektedir: 
nerede
C-skoru I-TASSER modellerin kalitesi ile güçlü bir korelasyon vardır. C-skoru ve protein süresini birleştirerek, ilk I-TASSER modellerinin doğruluğunu TM-skoru 2 ve RMSD 15 Å 0.08 ortalama bir hata ile tahmin edilebilir. C-skoru> modellerde - 1.5 Genel olarak, doğru bir kat olması beklenmektedir. Burada, RMSD ve TM-skoru her iki model ve doğal yapısı arasındaki topolojik benzerlik bilinen önlemlerdir. TM-skoru valu[0, 1] es aralığı girişi, daha yüksek bir skor daha iyi bir yapıya maç 16,17 gösterir . Ancak alt sırada modelleri (yani 2. -5 inci modelleri), TM-skoru ve RMSD C-skoru korelasyon çok zayıftır (~ 0.5), ve mutlak model kaliteli güvenilir tahmini için kullanılamaz.
Ilk modeli I-TASSER simülasyonları her zaman iyi bir model midir? Bu sorunun cevabı, hedef türüne bağlıdır. Kolay hedefler için, ilk modeli genellikle en iyi modeli ve onun C-skoru genellikle modellerin geri kalanından daha çok daha yüksektir. Ancak, sabit hedefler için parçacığı önemli şablon isabet bulunmamış, ilk model mutlaka en iyi model değildir ve I-TASSER aslında en iyi şablon ve modelleri seçiminde güçlük çeker. Bu nedenle sabit hedefler için 5 model, analiz etmek ve onları deneysel bilgi ve biyolojik bilgiye dayalı seçmek için tavsiye edilir.
Fonksiyon beklenenictions: Son adım olarak, FG-MD üretilen nihai 3D modelleri, yani protein fonksiyonunun üç yönü tahmin etmek için kullanılır: a) Enzim Komisyonu (EC) 18 ve (b) Gen Ontoloji (GO) 19 hüküm ve ( c) küçük molekül ligandlar için bağlayıcı siteleri. Üç yönü, fonksiyonel yorumlara kofaktör, PDB bilinen yapı ve fonksiyonları ile küresel ve yerel benzerlik şablon proteinlerine dayalı proteinin işlevi tahmin için yeni bir yaklaşım kullanılarak üretilir. İlk olarak, tahmin edilen modellerin küresel topoloji 20 TM-align yapısal uyum programını kullanarak fonksiyonel şablon kitaplıklarında karşı eşleştirilir. Sonra, bir dizi hedef modelleri en benzer proteinlerin global yapısı benzerlik dayalı kütüphane, geniş bir yerel arama bağlayıcı aktif / site bölge yakınında yapı ve sıra benzerlik belirlemek için yapılır. Ortaya çıkan küresel ve yerel benzerlik skorları sıralamak için kullanılırşablon proteinleri (fonksiyonel homologları) ve en yüksek puanı alan hit dayalı ek açıklama (AK numaraları ve Gen Ontoloji 19 açısından) transferi. Benzer şekilde, ligand bağlanma yeri kalıntıları ve ligand bağlayıcı modunda üst puanlama fonksiyonu şablonları 9 bilinen ligand bağlayıcı site kalıntıları ile sorgu yerel uyum dayalı anlaşılmaktadır.
Fonksiyon kalitesi (AT ve GO vadeli), I-TASSER tahmin, küresel ve yerel sorgu ve şablon arasındaki benzerlik bir ölçüsüdür fonksiyonel homoloji skoru (FH-skoru) göre değerlendirilir ve şu şekilde tanımlanır: 
C-skoru ifadede tanımlanan olarak tahmin modeli kaliteli bir tahmindir. (2), TM-skoru küresel model ve şablon proteinleri arasındaki yapısal benzerlik önlemler; RMSD ali model ve şablon yapısı arasındaki yapısal hizalı TM-align 20 bölgede RMSDCov yapısal uyum (yani sorgu uzunluğuna bölünmesiyle yapısal hizalı kalıntılarının oranı) kapsama temsil eder; ID ali TM-align hizalama sıra kimlik. AK numarası öngörüleri için tahmini güven puan olarak hesaplanır, tanımlanmış bir yerel bölge içinde sorgu ve şablon arasındaki aktif site maç (ACM) değerlendirmek için bir dönem de içerir: 
N yerel alan içinde mevcut şablon kalıntılarının sayısını temsil eder, N ali hizalı sorgu şablonu kalıntı çiftlerinin sayısı, d ii hizalı artıkları i th çifti arasında C α mesafe d 0 = 3.0 Å mesafe kesme, K ii hizalı kalıntılarının ITH çifti arasında BLOSUM puanları. Genel olarak, Fh-puan aralığı [0, 5] ve ACM [arasında 0, 2 puan, Daha güvende fonksiyonel atamaları gösteren yüksek puan. ACM skor aynı zamanda yerel yapısını ve BS-skoru olarak adlandırılan ligand bağlayıcı siteleri, yakın benzerlik dizisi değerlendirmek için kullanılır.
1. Protein dizisi Sunulması
- I-TASSER web sayfasını ziyaret edin http://zhanglab.ccmb.med.umich.edu/I-TASSER, yapısı ve fonksiyonu modelleme deneyinin ile başlar.
- Kopya ve sağlanan forma amino asit dizisi yapıştırın veya "Gözat" butonuna tıklayarak doğrudan bilgisayarınıza yükleyin. I-TASSER Sunucu şu anda 1500 kalıntılarının dizileri kabul eder. 1500 artıkları daha uzun Proteinler genellikle çok etki alanı proteinler, ve I-TASSER göndermeden önce, ayrı ayrı etki bölünebilir tavsiye edilir.
- E-posta adresi (zorunlu) ve iş (isteğe bağlı) için bir ad verin.
- Kullanıcılar isteğe bağlı harici arası-res belirleyebilirsinizidue temas / mesafe koltuk, eklemek-ek bir şablon veya yapı modelleme işlemi sırasında bazı şablon proteinler hariç. "Tartışma" bölümünde bu seçenekleri kullanma hakkında daha fazla bilgi edinin.
- Dizisi göndermek için, "Çalıştır I-TASSER" butonuna tıklayın. Tarayıcı kullanıcı belirtilen bilgi, iş tanımlaması (İş ID) numarası ve iş tamamlandıktan sonra yatırılır bir web sayfasına bir bağlantı gösteren bir onay sayfası yönlendirilecektir. Kullanıcılar bu bağlantıyı yer imi veya ileride başvurmak üzere iş kimlik numarasını bir yere not edin.
2. Sonuçların mevcudiyeti
- I-TASSER kuyruk sayfası ziyaret http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php tarafından sunulan iş durumunu kontrol edin . Arama sekmesini tıklayın ve İş kimlik numarası veya sunulan iş arama sorgu sırası kullanın.
- Yapısı ve fonksiyonu ay sonradeling bittiğinde, tahmin edilen yapılar ve bir web-link e-posta bir uyarı içeren görüntü size gönderilecektir. Bu bağlantıyı tıklayın ya da sonuçlarını görüntülemek ve indirmek için Adım 1.5 bağlantısını yer imi açmak.
3. İkincil yapı ve solvent erişilebilirlik tahminlerde
- Sonuç sayfasının üstünde görüntülenen FASTA biçimlendirilmiş bir sorgu sırasını kontrol edin. Ek herhangi bir kısıtlama / şablon dizisi sunulması, kullanıcı tarafından belirtilen bilgileri gösteren bir web sayfasına bir bağlantı sırasında belirtilmiş ise de (Şekil 1A) görülebilir.
- Alfa sarmal (H), beta iplikçik (S) veya bobin (C) ve her bir kalıntı tahmin güven skoru (0 = düşük, 9 = yüksek) olarak gösterilen ikincil yapı tahmini inceleyin. Protein çekirdek bölge tahmin etmek için, düzenli ikincil yapı (H ya da S) tahminleri ile uzun menziller bölgeye bakın. Yapısal protein sınıfı ayrıca analiz ikincil yapılar elementlerin dağılımı dayalı olabilir. Albu yüzden, protein bobin elemanları uzun bölgelerinde genellikle yapılandırılmamış / düzensiz bölgeleri göstermektedir.
- Sorguda tespit gömülü ve solvent maruz kalan bölgelerde tahmin edilen solvent erişilebilirlik (Şekil 1C). Değerleri 0 (gömülü kalıntı) 9 (kalıntı maruz) solvent erişilebilirlik aralığı öngördü. Çözücü maruz kalan ve hidrofilik artıkları olan bölgelerde potansiyel hidrasyon / işlevsel siteler çoğunlukla gömülü kalıntılarını içeren Bölgesi, protein çekirdek bölgeyi tanımlamak için kullanılan olabilir.
4. Tersiyer yapı tahminlerde
- Interaktif bir Jmol uygulaması (Şekil 2) görüntülenir sorgu protein tahmin üçüncül yapıları, görüntülemek için aşağı doğru kaydırın. Sol uygulaması görüntülenen yapının görünümünü değiştirmek için tıklayın, belirli bir bölge içine zum, öngörülen modele özgü kalıntı türleri seçmek veya kalıntı-arası mesafeleri hesaplamak.
- Uzun yapılandırılmamış bölgelerin varlığı modelleri analiz edin. Bu regions genellikle protein düzensiz bölgelere karşılık veya şablon uyum eksikliği gösterir. Bu bölgeler genellikle N & bölge modelleme doğruluğunu geliştirmek C-terminalindeki modelleme sırasında düşük modelleme doğruluk ve bu bölgelerde kaldırarak var.
- "Download Modeli" linklere tıklayarak modeli PDB biçimlendirilmiş yapı dosyalarını indirin. Bu yapısal özellikleri daha fazla analiz için herhangi bir moleküler görüntüleme yazılımı (örneğin Pymol, RasMol vb) bu dosyaları açabilirsiniz.
- Tahmin edilen yapıların kalitesini tahmin yapı modelleme güven skoru (C puanı) analiz edin. C-skoru (Eşitlik 2) değerleri aralığında genellikle [-5, 2], neyin daha yüksek bir puan daha kaliteli bir model yansıtır. Tahmini TM-skoru ve ilk model RMSD "Model 1 Tahmini doğruluk" olarak gösterilir. TM-skoru RMSD daha topolojik değişikliklere daha duyarlı olduğu gibi uzun proteinler, TM-skoruna göre model kalitesini değerlendirmek için tavsiye edilir. < li> C-skoru analiz etmek için bağlantı, tüm modelleri küme küme boyutu ve yoğunluğu "C-skoru hakkında daha fazla bilgi" üzerine tıklayın. Tahmini TM puan ve RMSD alt sırada modelleri C-skoru güçlü TM puan veya RMSD ile ilişkili olmadığını, çünkü, sadece ilk I-TASSER modeli sunulmuştur. Alt sıralarda yer modeller Kalite kısmen yakın ortalama doğal yapısına büyük küme ve yüksek yoğunluklu modelleri neyin küme yoğunluğu ve ilk model göre küme boyutu göre değerlendirilecektir olabilir.
- Düşük C-skoru tahminlerde genellikle düşük doğruluk tahmini göstermektedir. Çoğu böyle durumlarda, sorgu protein kütüphanede iyi bir şablon yoksundur ve ab initio modelleme (yani 120 artıkları) aralığında ötesinde bir boyutu vardır. Bu gibi durumlarda, kullanıcıların ek bir mekansal koltuk için aramak ve I-TASSER modelleme (Tartışma bölümüne bakınız) geliştirmek için bunları kullanmak. Aynı zamanda (QUARK sunucu dizileri göndermek için teşvik edilmektedirQUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) saf ab initio modelleme için protein boyutu 200 kalıntılarının altında ise.
5. LOMETS hedef şablon hizalama
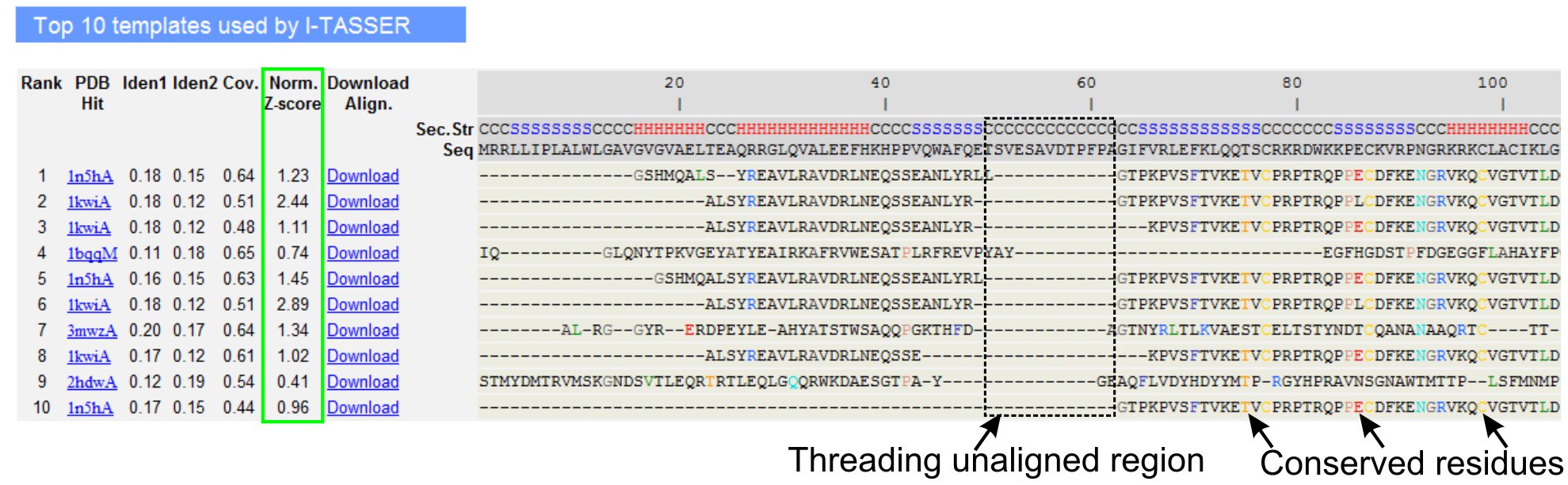
- LOMETS parçacığı programları (Şekil 3) tarafından belirlenen sorgu protein ilk on parçacığı şablonları, analiz etmek için aşağı doğru kaydırın. 'Norm gösterilen normalize Z-skoru (Eşitlik 1). Parçacığı hizalanmalar kalitesini analiz etmek için Z-skor 'sütunu. Normalleştirilmiş bir Z-skoru> 1 ile Hizalanmalar özgüvenli bir uyum yansıtır ve büyük olasılıkla sorgu protein olarak aynı kat var.
- Parçacığı Tabanlıdır bölgesi (sütun 'kimliklerinin sorgulandığını 1') ve sorgu ve şablon proteinler arasındaki homoloji değerlendirmek için tüm zincir (sütun 'kimliklerinin sorgulandığını 2') dizisi kimliğini analiz edin. Yüksek sıra kimlik sorgu ve şablon proteinler arasındaki evrimsel ilişkililik bir göstergesi.
- Görsel eksilerini belirlemek için renkli gösterilen parçacığı hizalı artıklarıerved artıkları / sorgu ve şablon proteinleri motifler. Parçacığı Tabanlıdır bölgedeki tüm zincir uyumuna göre yüksek sıra kimlik, ayrıca sorguda korunmuş yapısal motif / domains varlığını gösterir.
- Görüntüleyerek parçacığı uyum kapsama değerlendirin 'Cov' sütun ve hizalama teftiş. Üst hizalanmalar kapsamı düşük ve sorgu protein veya sorgu dizisi uzun bir segment için yok sadece küçük bir bölge ile sınırlı ise, daha sonra sorgu proteinin genellikle birden fazla etki alanı içerir ve sırası ve model ayırmak için tavsiye edilir ayrı ayrı etki alanları (Şekil 3).
- Üzerine tıklayarak PDB biçimlendirilmiş dizisi yapısı hizalama dosyaları indirin bağlantılar "Download Hizala". Malzeme bölümünde listelenen herhangi bir moleküler görüntüleme programında bu uyum dosyası açılacak ve yapı modelleme (Adım 1.4) ek koltuk eklemek için de kullanılabilir.
6.PDB de yapısal analogları
- 20 TM-align ilk tahmin modeli olarak tanımlanan ilk on yapısal analogları, yapısal uyum programı belirlemek için, sonuç sayfasında bir sonraki tablo (Şekil 4) . TM-skoru> 0.5 tespit analog ve model, TM-skoru <0.3 rastgele bir yapı benzerlik anlamına benzer bir topoloji var ve 16 sorgu protein yapısal sınıf / protein ailesini belirlemek için kullanılabilir gösterir.
- Dizisi kimliğini analiz ve yapısal olarak hizalanır bölgede RMSD gösterilen IDEN 've sütunlar yapısal analog modeli mekansal motiflerin korunması ve değerlendirilmesi için' RMSD '. Görme, bu yapısal olarak korunmuş kalıntıları ve motiflerle tanımlamak için hizalama renkli ve hizalı kalıntı çiftleri inceleyin.
- Yapısal sınıflandırma (SCOP hakkında RCSB web sitesini ziyaret edin ve daha fazlasını öğrenmek için 'Hit PDB' sütununda gösterilen PDB kodu tıklayınCATH ve PFAM) ve GO açısından ve bağlı ligand ilişkili fonksiyonel bilgiler (AK numarası).
7. Fonksiyon tahmin kullanarak kofaktör
- Sorgu protein fonksiyonel yorumlarını analiz sonuç sayfasında aşağı doğru kaydırın. Enzim Komisyonu (EC) numaraları, Gen Ontoloji (GO) açısından ve ligand bağlayıcı siteleri: Protein fonksiyonları görüntüleyen üç bağlamda tablolar numaralandırılır.
- 'TM-skoru', 'IDEN a' ve 'RMSD' Cov. ' her tablo sütunları global yapısı mekansal desen, model ve tanımlanan fonksiyonel homologları (şablonları) arasındaki benzerlik ve korunumu parametreleri analiz etmek.
8. Enzim Komisyonu numarası tahmini
- Gösterilen sorgu protein ilk beş potansiyel enzim homologları tablosu (Şekil 5) "AK numaraları Öngörülen". AK numarası tahmini bu şablonları kullanarak güven düzeyi 'EC-Puan' sütununda gösterilmiştir. Benchma üzerindegüvenilir bir şekilde analiz 23, sorgu ve şablon protein (AK numarası ilk 3 rakam) arasında işlevsel benzerlik rking AT-skoru> 1.1 kullanarak yorumlanabilir.
- Şablonları, sorgu protein olarak benzer kat (yani TM-skoru> 0.5) arasında görüş birliği (EC numaraları) fonksiyonu bakın. Eğer birden fazla şablonları aynı AK numarası ve AK-skoru> 1.1 varsa, tahmini güven düzeyi çok yüksektir. Ancak, AT-puanı yüksek ama tespit isabet arasında uzlaşma eksikliği varsa, o zaman tahmin daha az güvenilir hale gelir ve kullanıcılar GO vadeli tahminler danışmanız tavsiye edilir.
- Reaksiyonu katalize, co-faktör gereksinimlerine ve metabolik yolu da dahil olmak üzere ayrıntılı olarak şablon protein EXPASY Enzim veritabanını ziyaret edin ve fonksiyonunu analiz etmek için AK numaraları verilen linki, tıklayın.
9 - Gen Ontoloji (GO) vadeli tahminlerde
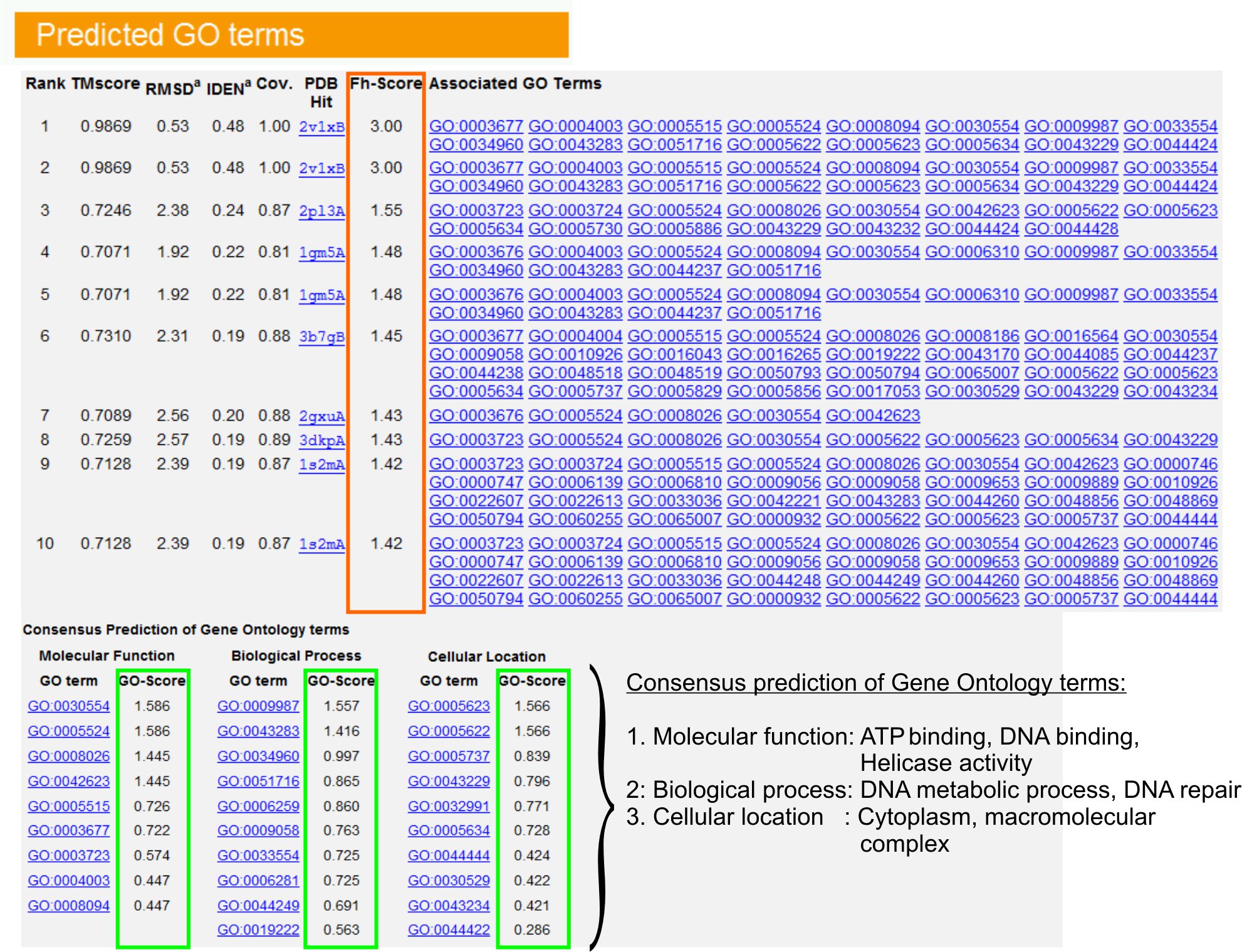
- "GO açısından Öngörülen" Görünüm tablosu (Şekilure 6) Gen Ontoloji (GO) açısından açıklamalı PDB kütüphane ilk on sorgu protein homologları, tespit etmek. Her protein genellikle moleküler fonksiyonları (MF), biyolojik süreçler (BP) ve hücresel bileşeni (CC) açıklayan çok GO şartları ile ilişkilidir. Amigo web sitesini ziyaret edin ve kendi tanım ve soy analiz etmek için her dönem tıklayın.
- Fh-skoru (Fonksiyonel homoloji puan) sütun, sorgu ve şablon proteinler arasında fonksiyonel benzerlik erişim ve bu proteinlerin fonksiyonel ek açıklama aktarılması güven düzeyi tahmin etmek için analiz edin. Bizim kıyaslama çalışmada 23,% 50 yerli GO açısından genel bir% 56 doğruluk ile 0.8 bir Fh-skoru kesme kullanarak ilk belirlenen şablon doğru tanımlanmalıdır.
- Fonksiyon şablonları arasında mutabakat analiz etmek için, "GO açısından fikir birliği tahmin" tablosu. Bu ortak fonksiyonları sorgu GO terimler (MF, BP ve CC) tahmin etmek için kullanılanprotein ve GO vadeli tahminlerin güven düzeyi (GO-skoru) değerlendirmek. Kıyaslama testi 23 dayanarak, en iyi yanlış pozitif ve yanlış negatif oranları, daha derin bir ontoloji düzeyde tahmin kapsama azalan, GO-skoru kesme tahminlerde = 0.5 için elde edilir .
10. Protein-ligand bağlanma yeri tahminlerde
- Ilk on sorgu protein ligand bağlayıcı site tahminlerde görmek için sayfanın altındaki aşağı doğru kaydırın. Öngörülen bağlayıcı siteleri ortak bağlayıcı cep paylaşan tahmin ligand konformasyonlarının sayısına göre sıralanır. Iyi tespit bağlanma yeri zaten Jmol uygulaması görüntülenir. Diğer öngörüleri analiz ve ligand etkileşim artıkları görselleştirmek için radyo düğmelerini tıklayın.
- BS-skoru sütun, model ve şablonun bağlayıcı sitesi arasında yerel benzerlik değerlendirmek için analiz edin. Kriter 9 dayanarak, BS-skoru> 1.1 yüksek sırası ve yapısı sim gösteririlarity model tahmin bağlama site ve şablon bilinen bağlanma yeri yakınındaki.
- "Download" linkine tıklayarak PDB biçimlendirilmiş yapı kompleksinin dosyasını indirin. Kullanıcılar herhangi bir moleküler görüntüleme programı bu dosyaları açmak ve etkileşimli tahmin bağlayıcı sitesi ve kendi yerel bilgisayarda ligand-protein etkileşimleri görebilirsiniz.
11. Temsilcisi sonuçları

Şekil 1-TASSER sonuç sayfasında bir alıntıyı gösteren (A) FASTA sorgu sırası biçimlendirilmiş; (B) ikincil yapısı ve buna bağlı güven puanları tahmin ve (C) kalıntılarının çözücü erişilebilirlik öngördü . Analiz çekirdek bölge ve sorgu potansiyel hidrasyon sitesi sırasıyla, açık mavi ve kırmızı dikdörtgenler vurgulanır.

Şekil 2.

Şekil 3. I-TASSER sonuç sayfasının LOMETS 5 parçacığı programlar tarafından ilk on tespit parçacığı şablonlar ve doğrultma gösteren bir örnek. Parçacığı hizalanmalar bir değer> 1 özgüvenli bir uyum yansıtır normalize Z-skoru (yeşil vurgulanır), kalite özelliklerine göre değerlendirilecektir. Ilgili sorgu artıkları aynı şablonda Bağlantısızlar kalıntıları korunmuş kalıntısı / motifin varlığını göstermek için renk vurgulanır çoğu uyum eksikliği üst şablonları, birden çok etki alanına, sorgu protein ve hizalanmamış kalıntılarının varlığı etki alanı ilintileyici bölgelere karşılık gösterir. Şekil 3 tam boyutlu sürümünü görmek için buraya tıklayın.

Şekil 4 tarafından belirlenen ilk on belirlenen yapısal analogları ve yapısal hizalamaları, sonuç sayfasında gösteren bir örnek 20 yapısal uyum programı TM-align. Gösterilen analogları sıralamasında yapısal uyum TM puan (mavi vurgulanmış) dayanmaktadır. TM-skoru <0.3 rasgele iki yapı arasında bir benzerlik anlamına gelir TM-skoru> 0.5, benzer bir topoloji karşılaştırıldığında iki yapıları olduğunu gösterir. "-" Hizalanmamış bölgeler ile gösterilir ise yapısal hizalı kalıntı çifti, amino asit mülkiyet dayalı renkte vurgulanır.ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Şekil 4 tam boyutlu sürümünü görmek için buraya tıklayın.

Şekil 5-TASSER sonucu sayfası PDB kütüphane sorgu protein tespit enzim homologları gösteren bir örnek. AK numarası tahmini güven düzeyi AT-AT-skoru> 1.1 sorgu ve şablon protein arasındaki fonksiyonel benzerlik gösterir (AK numarası aynı ilk 3 hanesi) skoru (yeşil vurgulanır) göre analiz edilir.

Şekil 6: I-TASSER sonuç sayfasında gösteren bir örnek sorgu protein vadeli tahminlerin GO . Gen Ontoloji şablon kütüphanesi sorgu protein Fonksiyonel homologları kendi Fh-skoru (turuncu dikdörtgen) göre sıralanır. Bu üst-puanlama hit Ortak işlevsel özellikleri otoma elde edilir sorgu protein son GO vadeli tahminlerin yedik. Tahmin GO açısından kalitesi tahmin GO-skoru (yeşil gösterildiği), GO-skoru> 0.5 güvenilir bir tahmin gösterir. Dayalı Şekil 6, tam boyutlu sürümünü görmek için buraya tıklayın.

Şekil 7-TASSER sonucu sayfası kofaktör 9 algoritma kullanarak ilk on proteinin ligand bağlanma yeri tahminlerin gösteren bir örnek. Tahmin edilen bağlayıcı sitelerin sıralaması sorgu ortak bağlayıcı cebinde payı tahmin ligand konformasyonlarının sayısına dayanır. BS-skoru (kırmızı vurgulanmış), yerel sırası ve tahmin edilen ve şablonun bağlanma yeri arasında yapı benzerliği bir ölçüsüdür; ve bağlayıcı site cepleri korunması analiz için yararlıdır.
les/ftp_upload/3259/3259fig8.jpg "/>
Şekil 8 kalıntı kalıntı temas / mesafe sınırlamalar belirtmek için kullanılan harici kısıtlama dosyaların bir örnek.

Şekil 9. Örnek bir şablon protein I-TASSER sunucusu belirtmek için kullanılan kısıtlama dosyaları. Veya (B) 3D formatında; (A) FASTA formatında ya da sorgu şablonu hizalama belirtebilirsiniz.

Şekil 10: I-TASSER yapı modelleme işlemi sırasında şablon hariç için kullanılan bir örnek dosya . İlk sütun hariç olmak için şablon proteinlerin PDB kimliği içerir. İkinci sütun, diğer benzer şablonları için gerekli olan şablon kütüphanesi kullanılacak sıra kimlik kesme belirtmek için kullanılır.
Discussion
Yukarıda sunulan protokol yapısı ve fonksiyonu modelleme I-TASSER sunucusu kullanarak için genel bir kılavuzdur. Bu otomatik işlem proteinlerin çoğu için çok iyi çalışır, rağmen, insan müdahaleler çoğu zaman özellikle yakın şablonlar PDB kütüphane eksikliği proteinler için önemli ölçüde modelleme doğruluğunu geliştirmek için yardımcı olur. (B) yapısı montaj geliştirmek için dış koltuk sağlayan; (a) çok etki alanı proteinler bölme ve (c) modelleme sırasında şablonları çıkarmadan: Kullanıcılar aşağıdaki şekillerde I-TASSER modelleme sırasında müdahalede bulunabilir.
Yarma çok etki alanı protein:
Birçok uzun protein dizileri, hem deneysel ve hesaplamalı teknikler kullanarak yapı aydınlatılması zorlaştırır esnek linker bölgeler, gergin birden çok etki alanına sık sık içerir. Bununla birlikte, etki alanları kişiler bağımsız katlama ve farklı moleküler işlevi gerçekleştirmek;ayrı ayrı etki, uzun multi-domain proteinleri ve model split istenebilir. Modelleme etki alanlarını ayrı ayrı sadece tahmin sürecini hızlandırmak değil, aynı zamanda sorgu-template uyum kaliteli, daha güvenilir bir yapı ve işlev tahminlerde artırır.
Protein dizileri etki alanı sınırları NCBI CDD 24, PFAM 25 veya InterProScan 26 gibi serbestçe kullanılabilir dış online programlar kullanılarak tahmin edilebilir. Ayrıca, sorgu protein LOMETS parçacığı hizalanmalar varsa, etki alanı sınırları görsel olarak (Adım 5.4) üst parçacığı şablonlar hizalanmamış artıkları uzun menziller belirlenmesi yer. Bu hizalanmamış bölgelerde daha çok etki alanı ilintileyici bölgelere karşılık gelir. Çok etki alanı şablonları şablon PDB hizalanmış tüm sorgu alanları ile kütüphanede mevcut olup olmadığını, daha sonra sorgu protein tam boy olarak modellenebilir.
Dış sınırlamalar sağlayın
A. kişi / mesafe koltuk belirtin
Deneysel karakterize arası kalıntı rehber / mesafeleri, NMR örneğin veyaçapraz bağlayarak deneyler, bir kısıtlama dosyasını yükleyerek belirtilebilir. Bir örnek dosya Şekil 8, Sütun 1 kısıtlama türünü belirtir gösterilir, "DIST" veya "İLETİŞİM" yani. Mesafe kısıtlama (DIST), sütunlar, 2 ve 4 kalıntı pozisyonlar (i, j) içeren, 3 ve 5 sütun belirtilen iki atomlar arasındaki mesafe belirten kalıntı ve sütun 6 atom türlerini içerir. Temas koltuk (İLETİŞİM), 2 ve 3 sütun iletişim halinde olmalıdır kalıntılarının pozisyonlar (i, j) içerir. Bu iletişime kalıntı çiftlerinin yan zincirleri merkezi arasındaki mesafe PDB bilinen yapılarda gözlenen mesafelerde göre karar verilir. I-TASSER yapısı arıtma simülasyonları sırasında belirtilen mesafe yakın bu atom çiftlerini çekmek için çalışacağız.
B. protein yapısı şablonu belirtin
LOMETS parçacığı programları sorgu Prot için makul kıvrımları bulmak için bir temsilci PDB kütüphane kullanımıein. Temsili bir yapısı kütüphanesi kullanarak dizisi yapısı hizalanmalar hesaplamak için gereken süreyi azaltmak için yardımcı olsa bile, bu, iyi bir şablonu protein, kütüphanede cevapsız veya şablon LOMETS parçacığı programlar tarafından tespit edilmiştir olmayabilir mümkündür kütüphanede mevcut. Bu durumlarda, kullanıcı şablonu olarak istenilen protein yapısı belirtmelisiniz.
Protein yapısı ek bir şablon olarak belirtmek için, kullanıcılar ya da bir PDB biçimlendirilmiş yapı dosyası yükleme veya PDB kütüphanede yatırılır protein yapısı PDB kimliği belirtin. I-TASSER Muster programı 23 kullanarak sorgu şablonu hizalama üretecek ve belirtilen kullanıcı şablon ve yapı montaj simülasyon kılavuzu LOMETS şablonları hem mekansal koltuk toplayacaktır. LOMETS koltuk doğruluğu farklı hedefler için farklı olduğundan, LOMETS koltuk ağırlık kolay (homolog) ta güçlüdürsistematik benchmark eğitim ayarlandı sabit (non-homolog) hedefler, daha rgets.
Kullanıcılar ayrıca, kendi sorgu şablonu hizalanmalar belirtebilirsiniz. : Sunucu, iki formatta uyum FASTA biçimi (Şekil 9A) ve 3D formatında (Şekil 9B) kabul eder. FASTA standart ve format adresinde açıklanan olduğunu http://zhanglab. ccmb.med.umich.edu / FASTA / . 3D formatı standart PDB formatı (benzer http://www.wwpdb.org/documentation/format32/sect9.html ), ancak şablonları türetilen iki ek sütunlar ATOM kayıtları (bkz. Şekil 9B) eklenir:
Sütunlar 1-30: Atom (sadece C-alfa) ve kalıntı sorgu sırası için isimleri.
Sütunlar 31-54: C-alfa şablonda ilgili atomlardan kopyalanan sorgu atomların koordinatları.
Sütunlar 55-59 Sorumlu kalıntı sayısı şablon dayalı hizalama
Sütunlar 60-64 Sorumlu kalıntı adı şablonu
Şablonlar proteinler dışla
Proteinler esnek moleküller ve biyolojik aktivitesini değiştirmek için birden fazla yapısal durumları kabul edebilir. Örneğin, birçok protein kinazlar ve membran proteinlerinin yapıları hem de aktif ve pasif yapısındaki çözüldü. Ayrıca bağlı ligand varlığı ya da yokluğu büyük yapısal hareketlere neden olabilir. Şablonun tüm yapısal durumları parçacığı programlar için benzer olsa da, sadece belirli bir devlet şablonları kullanarak sorgu modeli için arzu edilen bir durumdur. Sunucu üzerinde yeni bir seçenek, kullanıcı yapı modelleme sırasında şablon proteinler hariç sağlar. Bu özellik aynı zamanda kullanıcı modelleme için kullanılan şablonlar homoloji düzeyini seçmek için izin verecek. Kullanıcılar şablon proteinleri fr hariç tutabilirsinizom I-TASSER kütüphanesi:
A. bir dizi kimlik kesme belirtme
Kullanıcılar I-TASSER şablon kütüphanesi homolog proteinler dışlamak için bu seçeneği kullanabilirsiniz. Homoloji seviyesi dizisi kimlik limitine göre ayarlanır, yani sorgu ve sorgu sırası sıra uzunluğuna bölünmesiyle şablon protein arasında aynı kalıntı sayısı. Örneğin, eğer sağlanan formu "% 70" kullanıcı türleri,>% 70 sorgu protein I-I-TASSER şablon kütüphanesi dışında bir dizi kimliğe sahip tüm şablonları proteinler.
B. belirli bir şablon proteinleri Dahil Değildir
Belirli bir şablon proteinleri Dışlanacak yapıların PDB kimlikleri içeren bir liste yükleyerek I-TASSER şablon kütüphanesi dışında olabilir. Bir örnek dosya Şekil 10'da gösterilmiştir. Aynı protein PDB kütüphanede birden fazla giriş, I-TASSER se var gibirver varsayılan olarak belirtilen şablonları (Sütun1) yanı sıra bir kimlik kütüphane diğer tüm şablonları> belirtilen şablonlar% 90 hariç. Kullanıcılar ayrıca, kimliği ile tüm şablonları>% 70 belirli bir şablon proteinler dışı bırakılacaktır farklı bir kimlik kesim, örneğin% 70 belirleyebilirsiniz.
Disclosures
Çıkar çatışması ilan etti.
Acknowledgments
Proje Alfred P. Sloan Vakfı, NSF Kariyer Ödülü (DBI 1.027.394) ve Ulusal Genel Tıp Bilimleri Enstitüsü (GM083107, GM084222) tarafından kısmen desteklenmektedir.
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H.
- Finn, R. D.
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}