

Summary
Lignes directrices pour l'informatique basée caractérisation structurale et fonctionnelle de protéines en utilisant le pipeline I-Tasser est décrite. A partir de la séquence protéique de requête, les modèles 3D sont générés à l'aide de plusieurs alignements de filetage et itératif des simulations assemblage structural. Inférences fonctionnelles sont ensuite tirées à partir des matches à protéines avec une structure connue et fonctions.
Abstract
Génome projets de séquençage ont crypté des millions de séquences protéiques, qui exigent la connaissance de leur structure et leur fonction pour améliorer la compréhension de leur rôle biologique. Bien que les méthodes expérimentales peuvent fournir des informations détaillées pour une petite fraction de ces protéines, la modélisation computationnelle est nécessaire pour la majorité des molécules de protéines qui sont expérimentalement non caractérisés. Le serveur I-Tasser est une table de travail en ligne pour la modélisation à haute résolution de la structure des protéines et la fonction. Etant donné une séquence de protéine, une sortie typique du serveur I-Tasser comprend prédiction de structure secondaire, prédit accessibilité au solvant de chaque résidu, des protéines homologues modèle de threading et détectés par des alignements structure, jusqu'à cinq pleine longueur tertiaires modèles structurels, et basée sur la structure annotations fonctionnelles pour la classification des enzymes, des termes Gene Ontology et protéine-ligand des sites de liaison. Toutes les prévisions sont étiquetés avec un score de confiance quiraconte comment les prédictions sont précises sans connaître les données expérimentales. Afin de faciliter les demandes spéciales des utilisateurs finaux, le serveur fournit les canaux d'accepter spécifié par l'utilisateur inter-résidus de distance et de contact des cartes pour changer de façon interactive la modélisation I-Tasser, elle permet également aux utilisateurs de spécifier toutes les protéines de matrice, ou à exclure n'importe quel modèle protéines lors des simulations de montage de structure. Les informations structurelles pourraient être recueillies par les utilisateurs sur la base des preuves expérimentales ou connaissances biologiques dans le but d'améliorer la qualité de I-Tasser prédictions. Le serveur a été évalué comme le meilleur des programmes de la structure des protéines et des prévisions de fonction dans les expériences récentes CASP échelle de la communauté. Il n'y a actuellement> 20 000 chercheurs inscrits dans plus de 100 pays qui utilisent la ligne I-Tasser serveur.
Protocol
Méthode aperçu
Suivant le paradigme séquence à la structure à la fonction, la procédure I-Tasser 1-4 pour plus de la structure et la fonction de modélisation comporte quatre étapes consécutives suivantes: identification de modèle (a) par LOMETS 5; structure de fragment (b) le remontage par des répliques- l'échange des simulations Monte Carlo 6; (c) affiner la structure atomique niveau en utilisant REMO 7 et 8 FG-MD et (d) la fonction d'interprétations basées sur la structure en utilisant cofacteur 9.
L'identification Modèle: Pour une séquence de requêtes soumises par l'utilisateur, la séquence est d'abord enfilé à travers une bibliothèque structure de l'APB représentative par un LOMETS installés localement méta-threading du serveur. Threading est une procédure d'alignement de séquence-structure utilisée pour l'identification des protéines de modèle qui peuvent avoir une structure similaire ou contenir similaires motif structural que la protéine requête. Pour augmenter la couverture des homologues templdétections mangé, LOMETS combine plusieurs state-of-the-art algorithmes couvrant différentes méthodologies threading. Depuis les différents programmes de filetage ont des systèmes de notation différents et des sensibilités d'alignement, de la qualité des alignements générés enfilage de chaque programme est évalué par filetage normalisé Z-score, qui est défini comme suit: 
où Z-score est le score en unités d'écart type par rapport à la moyenne statistique de tous les alignements générés par le programme, et Z 0 est un programme spécifique de Z-score de coupure déterminé sur la base essais à grande échelle de référence de filetage 5 à différencier «bon »et« mauvais »modèles. Un modèle avec un haut Z-score signifie que les modèles haut ont un score d'alignement nettement plus élevé que la plupart des autres modèles, ce qui implique généralement que l'alignement correspond à un bon modèle. Si la plupart des modèles haut de filetage ont Salutnormalisée gh Z-scores, la précision de la finale, je Tasser-modèle est généralement élevé. Toutefois, si la protéine est grande et la couverture des alignements de filetage est confiné à une petite région de la protéine de requête, une grande normalisée Z-score ne signifie pas nécessairement une grande précision de la modélisation pour le modèle de pleine longueur. Haut deux alignements de filetage de chaque programme de filetage sont recueillies et utilisées pour la prochaine étape de l'assemblage de la structure.
Itérative de simulation de montage de structure: Suite à la procédure de filetage, séquence requête est divisé en régions filetage aligné et non aligné. Fragments continus dans l'alignement de filetage sont excisées à partir de modèles et utilisés directement pour l'assemblage de la structure, tandis que les régions de boucle non alignés sont construits par modélisation ab initio. La procédure d'assemblage structure est réalisée sur un système de treillis guidé par les simulations de Monte Carlo répliques d'échange 6. Le champ I-Tasser la force comprend l'hydrogène-bonding interactions 10, basée sur la connaissance termes d'énergie statistiques issues de structures de protéines connues dans l'APB 11, les prévisions basée sur une séquence de contact de SVMSEQ 12, et des contraintes spatiales recueillies auprès LOMETS 5 modèles threading. Les leurres de conformation générée dans les répliques à basse température au cours des simulations sont regroupées par Spicker 13 pour identifier les structures de faible énergie libre états. Centroïdes des grappes de grappes dessus sont obtenus en faisant la moyenne des coordonnées 3D de tous les leurres regroupés structurelle et utilisée pour la génération du modèle final. La simulation et la procédure de clustering sont répétées deux fois pour enlever les affrontements stérique et affiner la topologie globale.
Construction du modèle atomique de niveau et de raffinement: Les centroïdes pôle obtenue après regroupement Spicker sont des modèles réduits de protéines (chaque résidu représenté par son α C et la chaîne latérale du centre de masse) et have une application limitée biologique. La construction de plein atomiques modèle à partir de modèles réduits se fait en deux étapes. Dans la première étape, REMO 7 est utilisé pour construire des modèles atomiques plein à partir de C-alpha traces en optimisant les réseaux de liaison H. Dans la deuxième étape, REMO plein atomiques modèles sont affinés par FG-14 MD, ce qui améliore les angles de torsion dorsale, des longueurs de liaison, et les orientations rotamère chaîne latérale, par simulations de dynamique moléculaire, comme guidé par la recherche des fragments structuraux de la structures PDB par TM-align. Les modèles FG-MD raffinés sont utilisés comme modèles définitifs pour les prédictions de structure tertiaire par I-Tasser.
La qualité des modèles générés sont estimés en fonction d'un score de confiance (C-score), qui est défini sur la base du Z-score des alignements LOMETS threading et la convergence de I-Tasser simulations, mathématiquement formulé comme suit: 
où
Le C-score a une forte corrélation avec la qualité des modèles I-Tasser. En combinant C-score et la longueur des protéines, la précision de la première I-Tasser modèles peuvent être estimés avec une erreur moyenne de 0,08 pour le MC-score et de 2 A pour les 15 RMSD. En général, les modèles avec C-score> - 1,5 sont censés avoir un pli correct. Ici, RMSD et TM-score sont des mesures bien connues de similitude topologique entre le modèle et la structure native. TM-score Valugamme ES dans [0, 1], où un score plus élevé indique une meilleure structure de match de 16,17. Toutefois, pour les modèles de rang inférieur (soit 2 modèles ème ème -5), la corrélation de C-score avec TM-score et RMSD est beaucoup plus faible (~ 0,5), et ne peuvent pas être utilisées pour l'estimation fiable de la qualité du modèle absolu.
Est le premier modèle toujours le meilleur modèle dans I-Tasser simulations? La réponse à cette question dépend du type de cible. Pour des cibles faciles, le premier modèle est habituellement le meilleur modèle et son C-score est généralement beaucoup plus élevé que le reste des modèles. Toutefois, pour des cibles dures, où le filetage n'a pas de modèle coups importants, le premier modèle n'est pas nécessairement le meilleur modèle et I-Tasser a effectivement des difficultés à sélectionner le meilleur modèle et des modèles. Il est donc recommandé d'analyser toutes les 5 modèles pour cibles dures et les sélectionner sur la base des informations expérimentales et des connaissances biologiques.
Fonction predictions: Dans la dernière étape, finale modèles 3D générés par FG-MD sont utilisés pour prédire trois aspects de la fonction des protéines, à savoir: a) Enzyme Commission (CE) numéros 18 et Gene Ontology (b) (GO) 19 termes et ( c) les sites de liaison pour les ligands de petites molécules. Pour tous les trois aspects, les interprétations fonctionnelles sont générées en utilisant cofacteur, qui est une nouvelle approche pour prédire la fonction des protéines est basé sur la similarité globale et locale aux protéines de modèle dans l'APB avec une structure connue et fonctions. Tout d'abord, la topologie globale des modèles prédit est comparée des bibliothèques de modèles fonctionnels en utilisant le programme d'alignement structurel TM-align 20. Ensuite, un ensemble de protéines les plus similaires aux modèles cibles sont sélectionnés dans la bibliothèque en fonction de leur similitude structure globale, et une vaste recherche locale est effectuée pour identifier les similitudes structure et la séquence près de la région du site actif / contraignant. Les scores de similarité résultante globale et locale sont utilisés pour classer lesprotéines gabarit (homologues fonctionnels) et le transfert de l'annotation (numéros CE et Gene Ontology 19 termes) sur la base des coups obtenu les meilleurs résultats. De même, les résidus du site de liaison du ligand et le mode de fixation du ligand sont inférées basée sur l'alignement local de requête avec des résidus de ligand connu site de liaison dans les modèles haut fonction de score 9.
La qualité de la fonction (CE et GO terme) la prévision de l'I-Tasser est évaluée selon le score d'homologie fonctionnelle (FH-score) qui est une mesure de similarité globale et locale entre la requête et modèle, et est défini comme suit: 
où C-score est une estimation de la qualité du modèle prédit que défini dans l'équation. (2); TM-score mesure la similarité structurelle globale entre le modèle et les protéines de modèle; RMSD Ali est le RMSD entre le modèle et la structure de modèle dans la région structurellement alignées à partir de TM-align 20; Cov représente la couverture de l'alignement structurel (c'est à dire le rapport entre les résidus structurellement alignées, divisée par la longueur des requêtes); ID Ali est l'identité de séquence dans l'alignement TM-align. Le score de confiance pour les prévisions estimées numéro CE comporte aussi un terme pour évaluer match de site actif (ACM) entre requête et modèle dans une région définie locales, calculé comme suit: 
où N t représente le nombre de résidus de modèle présents dans la zone locale, N Ali est le nombre des paires alignées résidus requête modèle, D II est la distance entre les paires α C i e de résidus alignés, d 0 = 3,0 Å est la fréquence de coupure à distance, M II est des scores entre les deux BLOSUM vec des résidus alignés. En général, le FH-score est dans l'intervalle [0, 5] et le score ACM est entre [0, 2], Où des scores plus élevés indiquant une plus grande confiance missions fonctionnelles. Score ACM est également utilisé pour évaluer la structure locale et similarité de séquence à proximité des sites de liaison du ligand, ce qui est appelé BS-score.
1. Soumission de séquences de protéines
- Visitez la page Web I-Tasser au http://zhanglab.ccmb.med.umich.edu/I-TASSER à commencer par la structure et expérience de modélisation de fonction.
- Copiez et collez la séquence d'acides aminés dans le formulaire fourni ou directement les télécharger depuis votre ordinateur en cliquant sur le bouton "Parcourir". I-Tasser le serveur accepte actuellement des séquences avec un maximum de 1500 résidus. Protéines plus de 1500 résidus sont généralement multi-domaine des protéines, et sont recommandés pour être divisé en domaines individuels avant de soumettre à l'I-Tasser.
- Fournissez votre adresse e-mail (obligatoire) et un nom pour le poste (en option).
- Les utilisateurs peuvent éventuellement spécifier externes inter-rescontactez idue / distance de retenue, ajoutez-en un modèle supplémentaire ou d'exclure certaines protéines modèle lors du processus de modélisation de structure. En savoir plus sur l'utilisation de ces options dans la section "Discussion".
- Pour soumettre la séquence, cliquez sur "Exécuter I-Tasser" bouton. Le navigateur sera dirigé vers une page de confirmation de l'affichage des informations d'utilisateur spécifié, l'identification d'emploi (Job ID) numéro et un lien vers une page web où les résultats seront déposés après l'achèvement du travail. Les utilisateurs peuvent signet ce lien ou noter le numéro d'identification du travail pour référence future.
2. Disponibilité des résultats
- Vérifiez l'état de votre travail soumis en visitant la page file d'attente I-Tasser au http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Cliquez sur l'onglet recherche et utiliser le numéro d'identification d'emploi ou la séquence de requête pour rechercher votre travail soumis.
- Après la structure et la fonction moDeling est terminé, une notification par e-mail contenant l'image des structures prédites et une web-lien sera envoyé pour vous. Cliquez sur ce lien ou ouvrir le lien signet à l'étape 1.5 pour visualiser et télécharger les résultats.
3. La structure secondaire et les prédictions accessibilité au solvant
- Vérifiez la séquence requête FASTA formaté affiché sur le haut de la page de résultat. Si aucune restriction supplémentaire / modèle a été spécifié lors de la soumission de séquences, un lien vers la page Web affichant spécifié par l'utilisateur des informations peut aussi être vu (figure 1A).
- Examinez la prédiction de structure secondaire affiché comme: hélice alpha (H), les brins bêta (S) ou bobine (C) et score de confiance de la prévision (0 = faible, 9 = élevé) pour chaque résidu. Cherchez région avec de longs tronçons de la structure secondaire régulière (H ou S) les prévisions, pour estimer le noyau-région dans la protéine. Classe structurale de protéines peuvent également être analysées en fonction de la répartition des éléments de structures secondaires. Alainsi, les régions de longues éléments de l'antenne dans la protéine indiquent généralement des régions non structurées / désordonné.
- Voir l'accessibilité prédit solvant (figure 1C) de vérifier enterrés et solvant régions exposées dans la requête. Valeurs de la valeur prédite large accessibilité au solvant de 0 (résidus enfouis) à 9 (résidus exposés). Région contenant des résidus essentiellement enterrés peuvent être utilisés pour délimiter la région centrale de la protéine, alors que les régions avec un solvant de résidus hydrophiles sont exposées et l'hydratation potentiels / sites fonctionnels.
4. Prédictions de structure tertiaire
- Faites défiler pour afficher les structures tertiaires de protéines prédit requête, s'affiche dans une applet Jmol interactif (figure 2). Un clic gauche sur l'applet pour changer l'apparence de la structure apparaît, zoomer sur la région spécifique, sélectionnez les types de résidus spécifiques dans le modèle a prédit ou de calculer des distances inter-résidus.
- Analyser les modèles de la présence de longues régions non structurées. Ces rEGIONS correspondent généralement à des régions désordonnées dans les protéines ou indiquer un manque d'alignement modèle. Ces régions ont généralement faible précision de la modélisation et la suppression de ces régions lors de la modélisation de N & C-terminale région permettra d'améliorer la précision de la modélisation.
- Télécharger les fichiers PDB la structure du modèle formaté en cliquant sur le "Download Modèle" liens. Vous pouvez ouvrir ces fichiers dans n'importe quel logiciel de visualisation moléculaire (p. ex pymol, Rasmol etc) pour une analyse approfondie des caractéristiques structurelles.
- Analyser le score de confiance (C-score) de modélisation de structures d'estimer la qualité des structures prédites. C-Note (éq. 2) Les valeurs sont typiquement dans la gamme [-5, 2], dans laquelle un score plus élevé reflète un modèle de meilleure qualité. La TM-estimé score et RMSD du premier modèle est présenté comme "la précision estimée du modèle 1". Pour les protéines de long, il est recommandé d'évaluer la qualité du modèle basé sur TM-score, comme TM-score est plus sensible aux changements topologiques que RMSD. < li> Cliquer sur "plus sur le C-score" lien d'analyser C-partition, la taille des clusters et de la densité du cluster de tous les modèles. Estimation TM-score et RMSD ne sont présentés que pour la première fois que j'ai Tasser-modèle, car C-score de la baisse des modèles classés ne sont pas fortement corrélées avec TM-score ou RMSD. Qualité de rang inférieur modèles peuvent être partiellement évalués en fonction de leur densité et la taille du cluster grappe par rapport au premier modèle, où les modèles des plus grands clusters et une densité plus élevée sont en moyenne plus proche de la structure native.
- Faible C-score prédictions indiquent généralement une prédiction faible précision. Dans la plupart des cas, la protéine requête n'a pas un bon modèle dans la bibliothèque et a une taille au-delà de la gamme de modélisation ab initio (c'est à dire> 120 résidus). Dans ces cas, les utilisateurs peuvent rechercher des appuis supplémentaires spatiales et de les utiliser pour améliorer la modélisation I-Tasser (voir la section Discussion). Il est également encouragés à soumettre les séquences à notre serveur Quark (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) pour une modélisation ab initio pure si la taille des protéines est inférieure à 200 résidus.
5. L'alignement LOMETS matrice cible
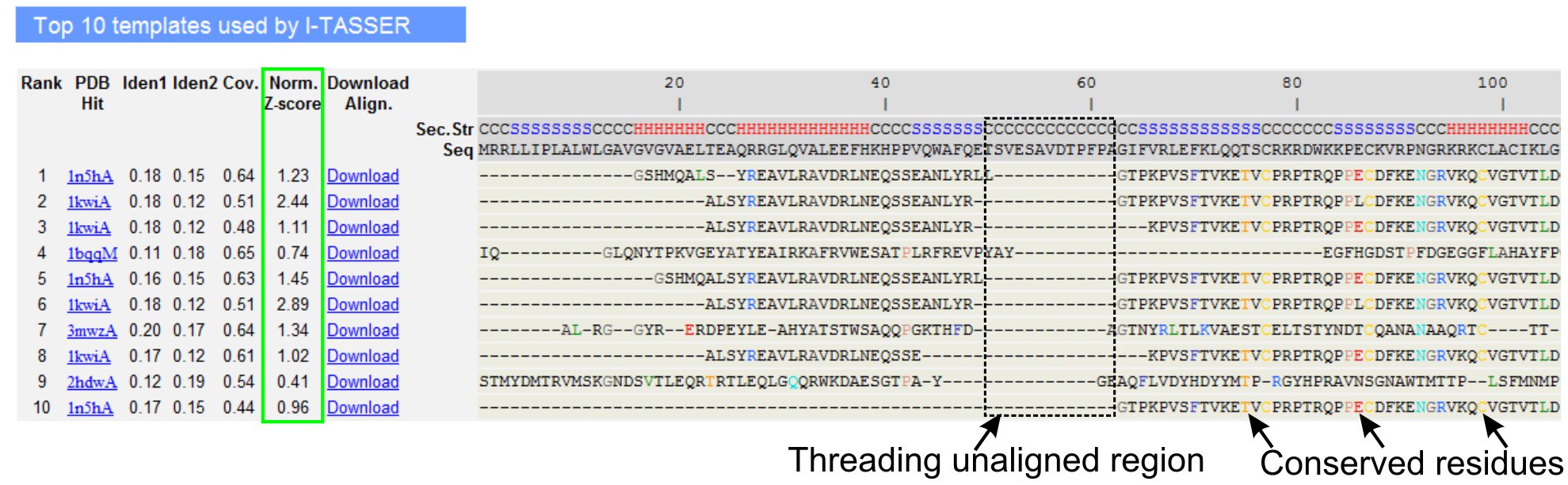
- Descendez à analyser les dix premiers modèles de threading de la protéine requête, comme identifié par les programmes LOMETS filetage (figure 3). Voir le Z-score normalisé (Eq. 1), illustré à la «norme. Z-score 'colonne, pour analyser la qualité des alignements de filetage. Alignements avec un Z-score normalisé> 1 reflète un alignement confiant et très probablement même pli que la protéine requête.
- Analyser l'identité de séquence dans la région de filetage-alignés ('Iden. 1' colonne) et pour toute la chaîne («Iden. 2 'colonne) afin d'évaluer l'homologie entre la requête et les protéines modèle. Identité de séquence élevé est un indicateur de parenté évolutive entre la requête et de protéines modèle.
- Voir les résidus de filetage alignées montré dans la couleur pour identifier visuellement des inconvénientsrésidus erved / motifs de la requête et les protéines modèle. Une identité de séquence plus dans le filetage alignés région, comparativement à l'ensemble de la chaîne d'alignement indique également la présence du motif structural conservé / domaines dans la requête.
- Évaluer la couverture de l'alignement de filetage en consultant le «Cov. colonne et inspecter l'alignement. Si la couverture des alignements supérieur est faible et limitée à seulement une petite région de la protéine requête ou absent pour un long segment de la séquence de requête, puis la protéine requête contient généralement plus d'un domaine et il est recommandé de fractionner la séquence et le modèle les domaines individuellement (figure 3).
- Télécharger l'APB fichiers au format d'alignement séquence-structure en cliquant sur le "Download Aligner" liens. Ces fichiers d'alignement peut être ouvert dans n'importe quel programme de visualisation moléculaire énumérés dans la section Documents, et peut également être utilisé pour ajouter des contraintes supplémentaires lors de la modélisation de la structure (étape 1.4).
6.Analogues structurels dans l'APB
- Voir le tableau suivant (figure 4) de la page de résultat pour déterminer le top dix des analogues structuraux du premier modèle prédit, tels qu'identifiés par le programme d'alignement structurel TM-align 20. Un MC-score> 0,5 indique que la analogiques détectées et modèle ont une topologie similaire et peut être utilisé pour déterminer la structure de classe / famille des protéines de la protéine 16 requêtes, tandis que ceux avec TM-score <0,3 signifie une similitude structure aléatoire.
- Analyser l'identité de séquence et RMSD dans la région structurellement alignées montré dans 'IDEN a' et 'RMSD une «colonnes d'évaluer la conservation des motifs spatiale dans le modèle et l'analogue structurel. Inspectez les paires de résidus de couleur et alignées dans l'alignement afin d'identifier ces résidus conservés structurellement et motifs.
- Cliquez sur le code PDB montré dans la colonne 'Hit APB "pour visiter le site RCSB et en apprendre davantage au sujet de leur classification structurelle (SCOP, CATH et PFAM) et des informations fonctionnelles (numéro CE, associé GO termes et ligand lié).
7. Prédiction de la fonction en utilisant cofacteur
- Descendez dans la page de résultat pour analyser les interprétations fonctionnelles de la protéine requête. Fonctions des protéines sont énumérées dans trois tableaux contexte, l'affichage: Enzyme Commission (CE) chiffres, Gene Ontology (GO) les modalités, et de sites de liaison du ligand.
- Voir le «MC-score», «RMSD a ',' IDEN a 'et' Cov. colonnes de chaque table pour analyser les paramètres de similitude structure globale et la conservation des structures spatiales entre le modèle et identifié homologues fonctionnels (modèles).
8. Prédiction de l'enzyme nombre Commission
- Voir le top cinq homologues d'enzymes potentiels de la protéine requête indiqué dans le "prédit numéros CE" tableau (figure 5). Le niveau de confiance de la prévision du numéro CE à l'aide de ces modèles est indiquée dans la colonne «EC-Score». Basé sur benchmarking analyse 23, la similitude fonctionnelle (3 premiers chiffres du numéro CE) entre la requête et de protéines modèle peut être interprété de manière fiable en utilisant CE-score> 1.1.
- Cherchez un consensus de la fonction (les numéros CE) parmi les modèles qui ont le pli semblables (c.-TM-score> 0,5) que la protéine requête. Si plusieurs modèles ont le même numéro CE et CE-score> 1,1, le niveau de confiance de la prévision est très élevé. Toutefois, si le CE-score est élevé, mais il ya un manque de consensus parmi les hits identifiés, alors la prévision devient moins fiable et les utilisateurs sont invités à consulter les prévisions GO terme.
- Cliquez sur le lien fourni sur le nombre CE pour visiter la base de données Enzyme ExPASy et analyser les fonctions, y compris la réaction catalysée, le co-facteur de besoins et de la voie métabolique, de la protéine de modèle dans le détail.
9. Gene Ontology prédictions terme (GO)
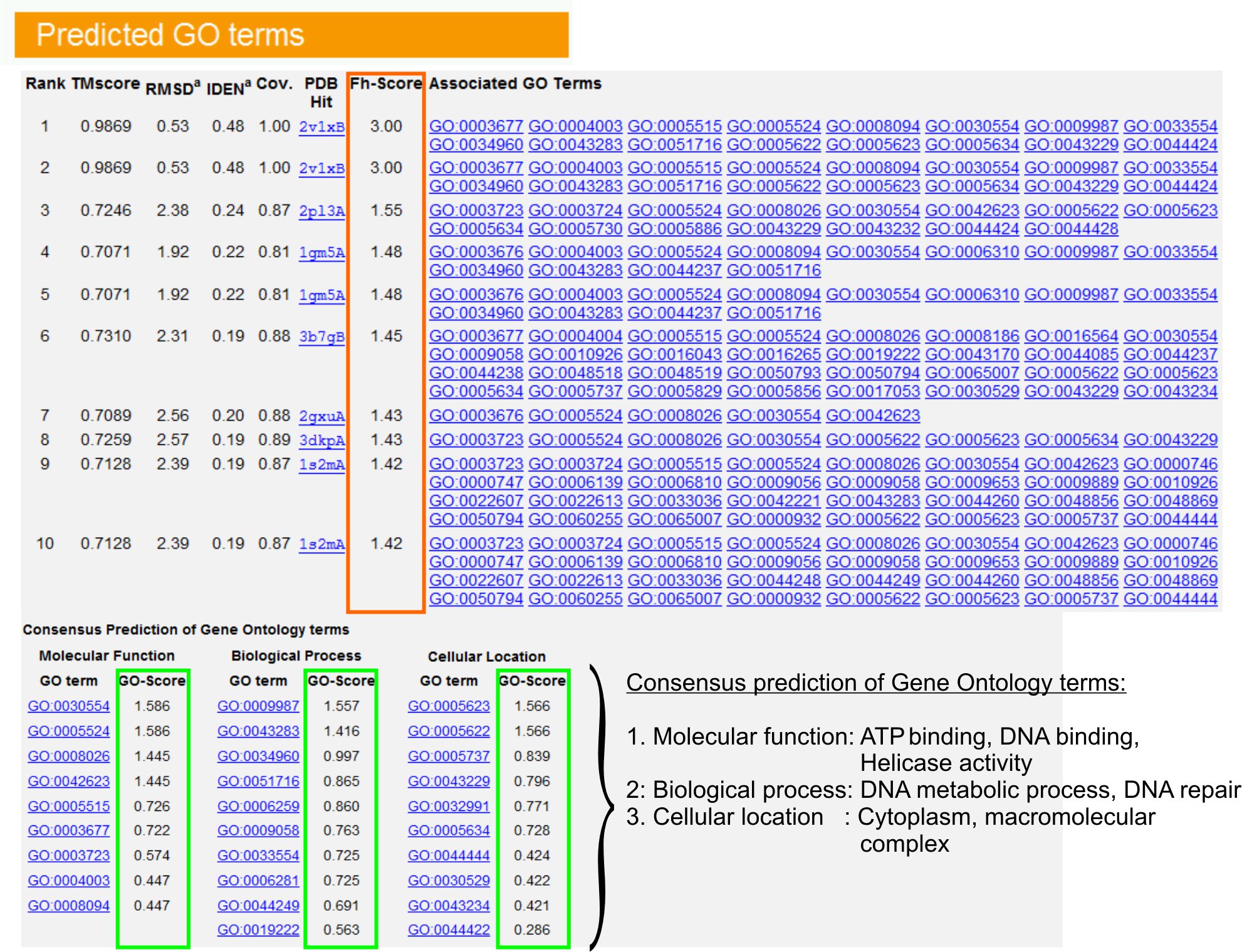
- Voir le "prédit GO termes« tableau (FigUre 6) pour identifier les dix premiers homologues de la protéine requête dans la bibliothèque APB, annoté avec Gene Ontology (GO) termes. Chaque protéine est généralement associée avec des termes GO multiples, décrivant ses fonctions moléculaires (MF), les processus biologiques (BP) et la composante cellulaire (CC). Cliquez sur chaque terme pour visiter le site Amigo et analyser sa définition et sa lignée.
- Analyser la colonne Fh-score (score d'homologie fonctionnelle) pour accéder à la similitude fonctionnelle entre la requête et des protéines de modèle et d'estimer le niveau de confiance de transférer l'annotation fonctionnelle de ces protéines. Dans notre étude comparative 23, 50% des termes GO natifs pourraient être correctement identifiés à partir du modèle d'abord identifié en utilisant une coupure FH-score de 0,8, avec une précision globale de 56%.
- Voir le "Consensus de prédiction des termes GO" tableau pour analyser l'assentiment de la fonction entre les modèles. Ces fonctions communes sont utilisées pour prédire les termes GO (MF, BP et CP) de la requêteprotéines et d'évaluer le niveau de confiance (GO-score) de GO prédictions terme. Basé sur le test de 23 benchmarking, le meilleur de faux positifs et faux négatifs sont obtenus pour des prédictions avec GO-seuil éliminatoire = 0,5, avec la diminution de la couverture de la prévision au niveau ontologique plus profond.
10. Protéine-ligand prédictions site de liaison
- Faites défiler la liste vers le bas de la page pour afficher les dix prédictions ligand site de liaison pour la protéine requête. Prévision des sites de liaison sont classés en fonction du nombre de conformations ligand prédit que l'action ordinaire poche de liaison. Le meilleur site identifié contraignante est déjà affichée dans l'applet Jmol. Cliquez sur les boutons radio pour analyser d'autres prédictions et de visualiser l'interaction des résidus ligand.
- Analyser la colonne BS-score pour évaluer la similarité locale entre le modèle et le site de liaison du template. Basé sur le point de repère 9, BS-score> 1,1 indique la séquence de haute et de la structure simsimilarité près du site prédit obligatoire dans le modèle et le site de liaison connue dans le modèle.
- Téléchargez le fichier de structure PDB formatés du complexe en cliquant sur le lien "Télécharger". Les utilisateurs peuvent ouvrir ces fichiers dans n'importe quel programme de visualisation moléculaire et interactivement visualiser le site prédit contraignantes et les interactions protéine-ligand sur leur ordinateur local.
11. Les résultats représentatifs

Figure 1 Un extrait de la page de résultat I-Tasser montrant (A) FASTA formaté séquence requête; (B). Prédit la structure secondaire et des scores de confiance associé, et (C) a prédit accessibilité au solvant des résidus. Région centrale et le site analysé le potentiel d'hydratation dans la requête sont surlignés en cyan et rectangles rouges, respectivement.

Figure 2.

Figure 3. Un exemple de page de résultat de I-Tasser montrant dix premiers modèles de threading et identifié par des alignements LOMETS 5 programmes de filetage. La qualité des alignements de filetage est évalué en fonction normalisée Z-score (surligné en vert), où une valeur> 1 reflète un alignement confiant. Résidus alignés dans le modèle qui sont identiques aux résidus requête correspondante sont surlignés en couleur pour indiquer la présence de résidus conservés / motif, tandis qu'un manque d'alignement dans la plupart des modèles haut indique la présence de plusieurs domaines dans la protéine requête et les résidus non alignés correspondent à des régions de liaison de domaine. Cliquez ici pour voir la version pleine grandeur de la figure 3.

Figure 4. Un exemple de page de résultat montrant dix identifié des analogues structuraux et des alignements de structure, identifiée par TM-align 20 programme d'alignement structurel. Le classement des analogues montré in est basé sur le TM-score (surligné en bleu) de l'alignement structurel. Un MC-score> 0,5 indique que les deux structures par rapport a une topologie similaire, tandis qu'un TM-score <0,3 signifie une similitude entre les deux structures aléatoires. Paires de résidus Structurellement aligné sont surlignés en couleur en fonction de leurs acides aminés de propriété, tandis que les régions non-alignés sont indiqués par «-».ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Cliquez ici pour voir la version pleine grandeur de la figure 4.

Figure 5. Un exemple de page de résultat de I-Tasser montrant homologues enzyme identifiée de la protéine requête dans la bibliothèque APB. Le niveau de confiance de la prévision du numéro CE est analysée en fonction CE-score (surligné en vert), où EC-score> 1,1 indique similitude fonctionnelle (même les 3 premiers chiffres du numéro de la CE), entre la requête et de la protéine modèle.

Figure 6. Un exemple de page de résultat de I-GO Tasser montrant prédictions terme pour la protéine requête. Homologues fonctionnels de la protéine requête dans la bibliothèque de modèles Gene Ontology sont classés en fonction de leur FH-score (dans le rectangle orange). Commune caractéristiques fonctionnelles de ces hits-de notation sont dérivées pour GENER mangeaient les prédictions finales terme GO pour la protéine requête. La qualité de l'prédit GO termes est estimé sur la base GO-score (en vert), où un GO-score> 0,5 indique une prédiction fiable. Cliquez ici pour voir la version pleine grandeur de la figure 6.

Figure 7. Un exemple de page de résultat de I-Tasser montrant dix protéines prédictions site de liaison du ligand à l'aide du cofacteur 9 algorithme. Le classement des sites prédit liaison est basé sur le nombre de conformations ligand prédit que l'action ordinaire poche de liaison dans la requête. BS-score (en rouge) est une mesure de séquence locale et la similitude de structure entre le prévu et le site de liaison du template, et est utile pour analyser la conservation des poches du site de liaison.
les/ftp_upload/3259/3259fig8.jpg "/>
Figure 8. Un exemple de fichiers de retenue externe utilisé pour spécifier des résidus de résidus de contact / contraintes de distance.

Figure 9. Exemple de fichiers de contrainte utilisés pour spécifier une protéine modèle pour le serveur I-Tasser. L'utilisateur peut spécifier l'alignement de requête-modèle, soit dans (A) format FASTA, ou (b) le format 3D.

Figure 10. Un exemple de fichier utilisé pour exclure modèle lors de la procédure I-Tasser la structure de modélisation. La première colonne contient l'ID de l'APB protéines modèle pour être exclu. La deuxième colonne est utilisée pour spécifier la fréquence de coupure d'identité de séquence qui sera utilisée pour d'autres modèles semblables dans la bibliothèque de modèles.
Discussion
Le protocole présenté ci-dessus est un guide général pour la structure et la fonction de modélisation utilisant le serveur I-Tasser. Bien que cette procédure automatisée fonctionne très bien pour la plupart des protéines, les interventions humaines contribuent souvent à améliorer significativement la précision de la modélisation, en particulier pour les protéines qui manquent de modèles proches de la bibliothèque APB. Les utilisateurs peuvent intervenir au cours de la modélisation I-Tasser de la manière suivante: (a) le fractionnement des protéines multi-domaines, (b) en fournissant contraintes externes pour améliorer l'assemblage de la structure, et (c) supprimer des modèles lors de la modélisation.
Fractionnement des protéines multi-domaines:
Beaucoup de séquences de protéines à long contiennent fréquemment de multiples domaines attachés par des flexibles régions linker, ce qui rend leur élucidation de la structure en utilisant des techniques difficiles à la fois expérimentale et computationnelle. Néanmoins, en tant que domaines sont indépendamment pliage des entités distinctes et peut effectuer la fonction moléculaire, il estsouhaitable de diviser longues protéines multi-domaines et le modèle de chaque domaine séparément. Domaines de modélisation individuelle ne sera pas seulement d'accélérer le processus de prédiction, mais augmente également la qualité de la requête-modèle de l'alignement, résultant dans la structure plus fiables et les prédictions de fonction.
Les limites du domaine dans les séquences de protéines peuvent être prédits à l'aide disponible gratuitement en ligne des programmes externes tels que NCBI CDD 24, 25 ou PFAM InterProScan 26. Aussi, si les alignements LOMETS filetage sont disponibles pour la protéine de requête, les limites du domaine peut être localisé par identifier visuellement de longues étendues de résidus non-alignés dans les modèles haut de filetage (voir l'étape 5.4). Ces régions non alignés pour la plupart correspondent à des régions de liaison de domaine. Si les modèles multi-domaines sont déjà disponibles dans la bibliothèque de modèles PDB avec tous les domaines requête alignées, alors la protéine requête peut être modélisée comme pleine longueur.
Fournir contraintes extérieures
A. Spécifier contact / distance de contraintes
Caractérisé expérimentalement inter-résidus contacts / distances, par exemple de la RMN ouréticulation des expériences, peut être spécifié en téléchargeant un fichier de retenue. Un fichier exemple est montré dans la figure 8, où la colonne 1 précise le type de contrainte, à savoir "DIST" ou "CONTACT". Pour de retenue à distance (DIST), les colonnes 2 et 4 contiennent des positions de résidus (i, j), les colonnes 3 et 5 contiennent l'atome-types dans les résidus et la colonne 6 indique la distance entre les deux atomes spécifiées. Pour les appuie-contact (contact), les colonnes 2 et 3 contiennent les positions (i, j) des résidus qui doivent être en contact. La distance entre les chaînes latérales au centre de ces paires de résidus en contact est décidé en fonction des distances observées dans les structures connues dans l'APB. I-Tasser va essayer de tirer de ces paires d'atomes proches de la distance spécifiée lors de l'affinement de la structure des simulations.
B. Spécifier un modèle de structure des protéines
LOMETS programmes filetage utiliser une bibliothèque représentant APB pour trouver les replis plausible pour la prot requêteein. Bien que l'utilisation d'une bibliothèque structure représentative contribue à réduire le temps nécessaire pour calculer les alignements de séquences-structure, il est possible qu'une protéine bon modèle est manquée dans la bibliothèque ou le modèle peut ne pas avoir été identifiés par les programmes LOMETS filetage, même si elle est présents dans la bibliothèque. Dans ces cas, l'utilisateur doit spécifier la structure de la protéine voulue comme le modèle.
Pour spécifier la structure des protéines comme un modèle supplémentaire, les utilisateurs peuvent soit télécharger un fichier de structure PDB formaté ou spécifier l'ID APB d'une structure de protéine déposée dans la bibliothèque de l'APB. Le I-Tasser va générer l'alignement des requêtes modèle à l'aide du programme 23 de rassemblement et permettra de recueillir contraintes spatiales à la fois du modèle de l'utilisateur spécifié et des modèles pour guider LOMETS la simulation de montage de structure. Parce que la précision des contraintes LOMETS est différente pour les différentes cibles, le poids des contraintes LOMETS est plus forte en facile (homologue) TArgets que celui dur (non homologues) des objectifs, qui ont été systématiquement à l'écoute de notre formation de référence.
L'utilisateur peut également spécifier leur propre requête template alignements. Le serveur accepte l'alignement en deux formats: le format FASTA (figure 9A) et le format 3D (Figure 9B). Le format FASTA est standard et décrite au http://zhanglab. ccmb.med.umich.edu / FASTA / . Le format 3D est similaire au format standard APB ( http://www.wwpdb.org/documentation/format32/sect9.html ), mais deux colonnes supplémentaires dérivées à partir des modèles sont ajoutés aux dossiers atome (voir figure 9B):
Colonnes 1-30: Atome (C-alpha uniquement) et les résidus de noms pour la séquence requête.
Colonnes 31-54: Les coordonnées de C-alpha des atomes de la requête copiés à partir des atomes correspondants dans le modèle.
Colonnes 55-59: numéro résidu correspondant dans le modèle basé sur l'alignement
Colonnes 60-64: nom résidu correspondant dans le modèle
Exclure les protéines modèles
Les protéines sont des molécules flexibles et peuvent adopter plusieurs états conformationnels de changer leur activité biologique. Par exemple, les structures des protéines kinases de nombreuses protéines membranaires ont été résolues dans la conformation à la fois actives et inactives. Aussi la présence ou l'absence de ligand lié peut provoquer de grands mouvements structurels. Alors que tous les états conformationnels du modèle sont semblables pour les programmes de filetage, il est souhaitable de modèle de la requête en utilisant des modèles dans un seul état particulier. Une nouvelle option sur le serveur permet à l'utilisateur d'exclure les protéines de modèle lors de la modélisation de structure. Cette fonction permettrait également à l'utilisateur de choisir le niveau d'homologie de modèles à utiliser pour la modélisation. Les utilisateurs peuvent exclure les protéines template from de la bibliothèque I-Tasser par:
A. Définition d'une coupure d'identité de séquence
Les utilisateurs peuvent utiliser cette option pour exclure des protéines homologues de la bibliothèque de modèles I-Tasser. Le niveau d'homologie est fixé en fonction du seuil d'identité de séquence, c'est à dire le nombre de résidus identiques entre la requête et la protéine modèle, divisé par la longueur de séquence de la séquence requête. Par exemple, si l'utilisateur tape dans "70%" dans la forme prévue, toutes les protéines modèles qui ont une identité de séquence> 70% à la protéine requête I-seront exclus de la bibliothèque de modèles I-Tasser.
B. Exclure protéines modèle spécifique
Protéines modèle spécifique peut être exclu de la bibliothèque de modèles I-Tasser en téléchargeant une liste contenant les ID APB des structures d'être exclus. Un fichier exemple est montré dans la figure 10. Comme la même protéine peut exister en tant que de multiples entrées dans la bibliothèque APB, I-Tasser soiRVer sera par défaut d'exclure les modèles spécifiés (dans Colonne1) ainsi que tous les autres modèles de la bibliothèque qui ont une identité> 90% aux modèles spécifiés. L'utilisateur peut également spécifier un seuil d'identité différents, par exemple, 70%, où tous les modèles avec l'identité> 70% aux protéines modèle spécifié seront exclus.
Disclosures
Pas de conflits d'intérêt déclarés.
Acknowledgments
Le projet est soutenu en partie par Alfred P. Sloan Foundation, NSF Career Award (DBI 1027394), et l'Institut national des sciences médicales générales (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}