

Summary
В данной статье описывается процедура подготовки флуоресцентно-меченных версия бактериофага лямбда-инфекции
Abstract
Система, состоящая из бактериофагов (фаг) лямбда и бактерии E. палочки долгое время служил в качестве парадигмы для клеточной судьбы определения 1,2. После одновременного заражения клетки ряда фагов, один из двух путей выбрано: литический (вирулентные) или лизогенных (спящих) 3,4. Мы недавно разработали метод флуоресцентной маркировки отдельных фагов, и смогли рассмотреть после заражения решения в режиме реального времени под микроскопом, на уровне отдельных клеток и фагов 5. Здесь мы описываем полный порядок осуществления инфекции экспериментов, описанных в нашей более ранней работе 5. Это включает в себя создание флуоресцентных фагов, заражение клеток, изображения под микроскопом и анализа данных. Флуоресцентные фага представляет собой «гибрид», совместно выразив дикого типа и YFP-фьюжн версий белков капсида ВВП. Неочищенного лизата фага впервые получены путем индукции lysogen из GPD-EYFP (EnhАНСЕД желтого флуоресцентного белка) фага, укрывательство плазмиды выражение дикого типа ВВП. Серия стадий очистки, затем выполняется, а затем DAPI маркировки и обработки изображений под микроскопом. Это делается для того, чтобы проверить равномерность, эффективность упаковки ДНК, сигнал флуоресценции и структурную стабильность фага акций. Первоначальный адсорбции фагов к бактериям осуществляется на льду, затем следует короткий инкубационный при 35 ° C, чтобы вызвать вирусную инъекцию ДНК 6. Фагов / бактерий смесь затем переехал на поверхность тонким питательным агаром плиты, покрытые покровным и отображаемого под эпифлуоресцентной микроскопом. После инфекционного процесса следует в течение 4 часов, в 10-минутного интервала. Несколько позиций этапе отслеживаются такие, что ~ 100 инфекциями клетки могут быть прослежены в одном эксперименте. В каждой точке координат и времени, изображений, приобретенных в фазового контраста и красные и зеленые флуоресцентные каналы. Фазового контраста изображения используется в дальнейшем для автоматической челл признания в то время как флуоресцентные каналы используются для характеристики инфекции результат: производство новых флуоресцентных фаги (зеленый), а затем лизиса клеток, или выражение лизогении факторов (красный), а затем возобновила рост и деление клеток. Полученные покадровой фильмы обрабатываются с помощью комбинации ручных и автоматических методов. Анализ данных результатов в выявлении инфекции параметров для каждой инфекции событие (например, количество и позиции заражения фагами), а также инфекции результат (лизис / лизогении). Дополнительные параметры могут быть извлечены при желании.
Protocol
1. Создание неочищенного лизата фага (рис. 1)
- В 50 мл колбу, привить новую колонию LE392 (λ LZ1) [pPLate * D] (см. Таблицу 1 для подробностей) в 6 мл LB среде 7 с добавлением 10 мкг / мл канамицина и 100 мкг / мл ампициллина. Расти в течение ночи при 30 ° C с мягким встряхиванием (180 оборотов в минуту).
- Развести культуры 1:100 в LBM (LB с добавлением 10 мМ MgSO 4) и растут при температуре 30 ° C с мягким встряхиванием (180 оборотов в минуту). Для того, чтобы оптимизировать выход фага, убедитесь, что культура объеме не более одной десятой части потенциала объем колбы. Как правило, мы подготовим два флакона 2-литровым или 2,5-литровым мощностью, и добавить 2,5 мл ночной культуры в 250 мл LBM среды в каждой колбе.
- Когда плотность клеток достигает OD 600 ≈ 0,6 (~ 2,5 - 3 часа), вызывают lysogen путем перемещения культуры в 42 ° С водяной бане шейкере в течение 18 минут с мягким встряхиванием (180 оборотов в минуту), а затем incubatе при 37 ° C с мягким встряхиванием (180 оборотов в минуту) до лизиса видно (культура становится ясно, в ~ 60 - 90 мин).
- Добавить 2% хлороформа к культуре, встряхните рукой, чтобы смешать, а затем инкубировать в течение 15 мин при комнатной температуре. Внимание: Надевайте перчатки для обработки хлороформом, и не вдыхайте его.
- Передача культуры в две 250-мл бутылки центрифуги, центрифуги культуры в Sorvall GSA ротора при 10000 оборотов в минуту в течение 15 мин при 4 ° C. Восстановление супернатанта, содержащего частицы фага, и отбросить осадок от мусора. Выполнение второго центрифугирования, чтобы убедиться, чтобы избавиться от видимого мусора.
- Используйте стандартный протокол титрования фага 8 до измерения концентрации фагов. Титр фага должно быть ~ 5-10 х 10 9 БОЕ / мл. Используйте supF деформации, таких как LE392 в качестве индикатора деформации из-за SAM7 мутации в генотипе флуоресцентных фагов, а также использовать верхний агар и агар пластин с богатым NZYM получить больше бляшек (Fiрисунке 2).
2. Фаг очистки (рис. 1)
- Залить лизат в большой (например, 2-литровый) колбу, добавить ДНКазой I и РНКазой (1 мкг / мл) в лизат для того, чтобы переварить нуклеиновых кислот, освобожденных из лизированных бактерий, и инкубировать 1 час при комнатной температуре.
- Добавить 1 М NaCl в лизат, передает лизат в 250 мл бутылки центрифуги, и инкубировать 3 часа на льду. Центрифуга лизат в Sorvall GSA при 10000 оборотов в минуту в течение 15 мин при 4 ° C. Восстановление супернатант. Титр фага должна быть такой же, сырой лизат, который составляет ~ 5-10 х 10 9 БОЕ / мл. Добавление NaCl способствует диссоциации частиц фага с бактериальной мусора и требуется для эффективного осаждения частиц фага с помощью ПЭГ 8.
- Залить лизат в большой колбе, например, 2-литровую колбу, добавьте 10% (вес / объем) PEG8000 в лизат, медленно перемешать или встряхнуть, чтобы растворить PEG8000 при комнатной температуре. Передача лизат на 250 процентов млrifuge бутылки, а затем инкубировать в течение ночи (~ 16 часов) при 4 ° C. Центрифуга лизат в Sorvall GSA ротора при 10000 оборотов в минуту в течение 15 мин при 4 ° C. Удалите супернатант.
- Замочите гранул (частиц фага осаждают PEG8000) с фагом SM буфера (4 мл SM буфер на 250 мл начальная лизата фага). Инкубируйте с очень мягким встряхиванием или нет встряхивания в течение 16 ч при 4 ° C.
- Осторожно вынуть лизат (SM буфера с фаговых частиц) в 50 мл центрифужные пробирки Eppendorf, а затем вымыть оставшиеся гранулы с 0,5 - 1 мл буфера SM.
- Добавить равным объемом хлороформа в лизата. Аккуратно перемешайте лизат с хлороформом путем обращения вверх и вниз несколько раз. Центрифуга при 4000 оборотов в минуту в течение 15 мин при 4 ° С в Eppendorf 5804R или аналогичный настольная центрифуга.
- Повторите Шаг 2,6 до получить более четкое лизата. Титр фага должно быть ~ 1-2 х 10 11 БОЕ / мл.
- Подготовка SM / CsCl решения с трех разных плотностей (ρ) 1,3 г / мл, 10,5 г / мл и 1,7 г / мл. Измерение показателя преломления (η), чтобы получить более точные показания плотности. Плотность преобразования 9, ρ = 10,8601 η - 13,4974 на 25 ° C. См. таблицу 3 для деталей.
- Используйте шприц с длинной иглой, чтобы загрузить раствора в 14 мл сверх-четкое Beckman ультрацентрифуге труб 40Ti. Чтобы избежать смешивания и сформировать лучшие градиента плотности, нижележащих решения (т.е. слои решений увеличением плотности под другом) должны быть использованы, например, аккуратно загрузить 2 мл SM / CsCl решений в порядке 1,3 г / мл, 1,5 г / мл и 1,7 мкг / мл, вставив иглу с 3 мл шприц в нижней части трубы.
- Аккуратно загрузите 8 мл фага лизат, накладывая сверху 14 мл ультрацентрифуге труб. Подготовка баланса трубку. Центрифуга в Beckman SW40Ti ротора при 24000 оборотов в минуту в течение 4 часов при температуре 4 ° C.
- Осторожно вынуть трубку в темную комнату и освещают из верхней части трубки на черном фоне в режиме автофокусаlashlight. Фагов группы должны быть ясно видны на месте интерфейса между 1,3 г / мл и 1,5 г / мл SM / CsCl слоев (рис. 3А). Прокол через боковые трубки чуть ниже полоса с 21,5 игла с 3 шприца мл. Аккуратно собираем ~ 500 мкл фага подвески. Титр фага должно быть ~ 5-10 х 10 11 БОЕ / мл.
- Поместите фага суспензии в 4 мл сверх-четкое Beckman ультрацентрифуге SW60Ti трубы ротора. Заполнить трубу с 1,5 г / мл SM / CsCl решение. Подготовка баланса трубку. Центрифуга в Beckman SW60Ti ротора при 35000 оборотов в минуту в течение 24 ч при 4 ° C.
- Повторите ту же процедуру, что и в шаге 2,11 собрать фага из видимого диапазона. Группа должна быть видна, как показано на рисунке 3b.
- Загрузите фага подвески в кассету диализной мембраны (табл. 2) и диализ три раза против 1000-кратного объема буфера SM при 4 ° С в течение продолжительность 3 часа, 3 часа и overnigHT (~ 16 часов). Цель диализа, чтобы избавиться от CsCl присутствует в фаг подвески. Окончательный титр фага должно быть ~ 5-10 х 10 11 БОЕ / мл.
3. Подготовьте агарозном геле плиты (рис. 4)
- Очистите 6 предметные стекла (75 х 50 мм, толщина 1 мм) с 70% этанола.
- Организовать 5 слайдов и закрепите ленту как показано на рисунке 4.
- Смешать 0,09 г агарозы в 6 мл среды в маленьком стакане, накрытом цепляются обертка (уступая 1,5% агарозном). Нагрейте на горячей плите, пока раствор не станет прозрачной.
- Залейте раствор агарозы на обеспечение слайдов.
- Установите последний слайд на вершине, тщательно избегая пузырьков воздуха. Место веса на вершине и дайте ему остыть в течение ~ 30 мин.
- Удалите 4 слайдов на стороне, и оберните плиту вместе с верхней и нижней слайды с цепляются обертка. Плиты можно хранить при температуре 4 ° С в течение 3 дней.
4. Тестирование очищенных фагов складе
- ГотовитьPBS-агарозном геле плиты, как описано выше (раздел 3).
- Пятно очищенных фагов с DAPI. Смешать 10 мкл фага (~ 1 х 10 10 БОЕ / мл) с 10 мкл 10 мкг / мл DAPI (конечная концентрация DAPI 5 мкг / мл), инкубировать в течение 30 мин при 4 ° С или 10 мин при комнатной температуре.
- Место 1 мкл фага / DAPI смесь в центр № 1 24 х 50 мм покровным, наложение небольшой кусок (~ 10 х 10 мм) из заранее подготовленных PBS-агарозы плиты. Небольшой кусок плиты агарозном разрезают бритвой после верхнего слайда на сэндвич-гель удаляется. Изображение образца под микроскопом эпифлуоресцентной через YFP и DAPI каналов. Индивидуальные фагов должно быть видно, как дифракционной флуоресцентных «пятен» в обоих каналах (рис. 5). Используйте тот же микроскоп и настроек камеры, как и в шаге 6.2.
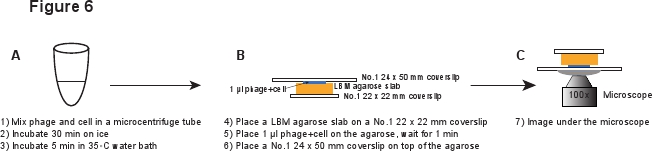
5. Инфекция (рис. 6)
- В 14 мл трубки Falcon, привить новую колонию LE392 [PP RE-MCHerry] (см. Таблицу 1 для подробностей) в 2 мл LB среде, содержащей 100 мкг / мл ампициллина, 10 мМ MgSO 4 и 0,2% мальтозы. Расти в течение ночи при 37 ° C при умеренном встряхивании (265 оборотов в минуту).
- Развести культуры 1:1000 в LBMM (LB с добавлением 10 мМ MgSO 4 и 0,2% мальтозы), т.е. добавить 5 мкл ночной культуры в 5 мл LBMM среду в 50 мл колбу. Вырасти до OD 600 ≈ 0,4 при 37 ° C при умеренном встряхивании (265 оборотов в минуту).
- Используйте LBM среде подготовить LBM-агарозном геле плиты, как описано выше в разделе 3.
- Центрифуга 1 мл клеток при 2000 г в настольный микроцентрифуге в течение 2 мин при комнатной температуре. Удалить супернатант и осторожно ресуспендируют клеток в 20 мкл ледяной LBMM для достижения OD 600 и 20.
- При манипулировании очищенных фагов акции, использовать широкий кончика пипетки или сократить регулярные кончика пипетки, чтобы сделать кончик открытия шире, чтобы избежать сдвига частиц фага 3. Аккуратно MIX 20 мкл клеток с 20 мкл очищенных фагов, чтобы достичь среднего фагов к клетке соотношение в диапазоне 0,1 - 5. Инкубировать на льду в течение 30 мин, чтобы адсорбции фага, а затем инкубировать в 35 ° С водяной бане в течение 5 мин, чтобы вызвать ДНК фага инъекций 6.
- Внесите вверх и вниз несколько раз, чтобы отделить любые клеточные агрегаты. Снова используем широкий кончика пипетки, чтобы избежать сдвига фагов. Развести смесь в 1:10 LBMM, например, 5 мкл смеси в 45 мкл LBMM.
- Поместите кусок LBM-агарозы плиты (~ 10 х 10 мм) на No.1 22 х 22 мм покровным. Заранее подготовленные LBM-агарозы плиты должны быть размещены при комнатной температуре в течение не менее 1 часа перед использованием, чтобы убедиться, что плита агарозном достигнет комнатной температуры. Место 1 мкл фага / смеси клеток на плите агарозы и ждать в течение 1 мин, чтобы смесь впитаться в плите агарозы. Аккуратно № 1 24 х 50 мм покровным на верхней плите агарозы. Эта процедура предназначена для предотвращения сдвига фагов с инфected ячейки (рис. 6).
6. После судьбы клеток под микроскопом
- Осторожно установите покровное на сцене микроскопом. Для визуализации используется большим увеличением (например, 100x) цели (см. Микроскоп системы в обсуждение ниже).
- Получить изображение, установленное для начального периода времени. Этот набор изображений будет использоваться для характеристики начального номера и позиции всех заражения фагами. Снимите серию из 15 фотографий при 200 нм оси (вертикальной) интервалы. Изображение через канал YFP. Кроме того, сделайте один в фокусе изображения с помощью фазового контраста и mCherry каналов. Оптимизация интенсивности света и времени экспозиции для получения достаточного сигнала при минимальных отбеливания и повреждение клеток (см. Image Acquisition в обсуждение ниже).
- Приобретать покадровой фильм после инфицирования клетки судьбу. Изображение образца в фазово-контрастной, YFP и mCherryканалов в интервалах времени от 10 мин около 4 часов. Во время покадровой фильм, использовать единую позицию г-изображения для каждого канала в момент времени, чтобы избежать ненужного воздействия на образец, который может привести к обесцвечиванию и фототоксичности.
7. Анализ изображений
- Вручную посчитать количество фагов и запись расположения фага и клетки длиной в начальный период времени. Это можно сделать с помощью программного обеспечения, таких как MetaMorph или ImageJ. Запись ячейки судьбы (литические, лизогенных или неинфицированных), лизис время, и любой другой нужной информации, играя в замедленную фильма. Для выявления различных клеточных судеб, см. замедленной фильм в разделе представителя Результаты ниже.
- В дополнение к ручной анализ выше, более количественную информацию (например, уровень флуоресценции с течением времени в отдельных клетках) может быть извлечено с помощью автоматизированного клеточного распознавания и отслеживания линии алгоритмов. Мы используем дома, построенного Matlab программа TRacing линии флуоресценции клеток и уровни, вместе с кодом Schnitzcell Matlab для сотовых сегментации (написана группой Elowitz в Калифорнийском технологическом институте).
8. Представитель результаты:
Фаг Покрытие:
Бляшки флуоресцентно меченных фагов (в шаге 1.6 и раздел 2) значительно меньше, чем у дикого типа (рис. 2). Поэтому мы инкубировать пластин по крайней мере 12 часов при 37 ° C инкубатора для бляшками, чтобы быть видимыми.
Фаг ультрацентрифугирования:
После ультрацентрифугирования фага образца градиента CsCl шага (шаг 2,10), две полосы должна быть видна (рис. 3А). Верхняя полоса, на границе между подвеской фага и SM / CsCl 1,3 г / мл слой, содержит мусор и пустые ячейки капсид фага. В нижней полосе, на границе между SM / CsCl 1,3 г / мл и 1,5 г / мл слоев, является фаг группы. Тхис группой появляется зеленоватый для флуоресцентных фага λ LZ2. Полоса для дикого типа фага λ IG2903 появляется голубовато 5. После ультрацентрифугирования CsCl градиент равновесию в шаге 2.12, одного фага группы должны быть видны в средней части трубы (рис. 3В). С люминесцентными фага λ LZ2 содержит смесь GPD-EYFP и ВВП капсид, соотношение белок-ДНК выше, чем у дикого типа. Таким образом, группа флуоресцентных фага λ LZ2 немного легче (кажется, на более высоком месте в трубке), чем у дикого типа λ IG290310.
DAPI окрашивания:
На рисунке 5 показаны типичные изображения, полученные после маркировки фага с DAPI (раздел 4). YFP и DAPI сигналы успешно очищенных фагов должно быть близка к 100% соответствия. Как правило, мы видим, что менее 1% пятна YFPы не содержат DAPI (представляющих капсид без вирусного генома). Менее 1% пятен DAPI не содержат YFP (соответствующий нефлуоресцентного фаги) 5.
Покадровый фильма:
Литические клетки признаны накопления YFP флуоресценции (зеленый канал) внутрь клетки, а затем лизиса клеток. Лизогенных клеток признаны накопления равномерное mCherry флуоресценции (красный) внутри клетки и возобновления нормального клеточного роста и деления. Неинфицированных клеток (или клеток, где инфекция не удалось) не будет отображать любую из фенотипов выше, и будет расти и делиться нормально. 7 показано несколько изображений множества фазового контраста, YFP и mCherry каналам, и соответствующие наложения изображений этих трех каналов, от обычной покадровой фильма (раздел 6). Отдельные фаги (зеленые пятна), хорошо видны на начальных сроков (рис. 7А). Как правило, числофагов видны на поверхности клетки (предположительно заражение этих клеток), а другие фаги неадсорбированный, как показано на рисунке 7Б (левая панель). Инфекции результатом становится различимым с течением времени. Литического цикла указывается внутриклеточных производство новых фагов (зеленый, рис. 7), а затем лизиса клеток (клеток с взорвался выпустил зеленые фагов, рис 7D). Лизогении указывается производства mCherry от промотора P RE (красный, рис 7С) и возобновление рост и деление клеток (красный, рис 7D).




Рисунок 1. AB). Фагов очищается через ряд шагов (панелей CL).

Рисунок 2. Фаг бляшек. Бляшки флуоресцентных фаг (слева) меньше, чем у дикого типа (справа) после инкубации пластин в течение 12 ч при 37 ° C.

Рисунок 3. Фаг полосы после ультрацентрифугирования.) Две полосы видны после центрифугирования в градиенте CsCl шаг. Верхний соответствует мусор и пустые ячейки капсид фага, нижняя полоса содержит необходимый фага. Слева: флуоресцентные фага, право. Дикого типа B ) Одной полосы фага видна после центрифугирования в градиенте CsCl равновесия. Флуоресцентные группы фагов (слева) зеленоватая, по сравнению с голубоватым группу для диких фаготипа (справа).

Рисунок 4. Порядок подготовки плит агарозном геле.

Рисунок 5. Флуоресцентные изображения фагов после окрашивания DAPI. Индивидуальные фаги легко различимы, и YFP и DAPI сигналы ко-локализуются очень хорошо.

Рисунок 6. Схематическое описание инфекции фагом и визуализации установки. Щелкните здесь для просмотра полноразмерной версии этого образа.
рисунке 7 "SRC =" / files/ftp_upload/3363/3363fig7.jpg "/>
Рисунок 7. Типичные изображения с покадровой фильм фаговой инфекции. Показаны фазового контраста, YFP и mCherry каналов, а также наложения из трех каналов. (A) YFP-изображения от первоначального срока. Слева, на сумму YFP изображений в различных Z-позиции. Три правом изображения образца YFP изображений в различных Z-позиции, соответствующие различным областям поверхности клетки. (B), (C) и (D) Наложение изображений (слева) фазового контраста (средний слева), YFP (средний справа) и mCherry (справа) каналов на разных временных рамках. (B) при Т = 0, две клетки видны, каждый зараженный одним фагом (зеленые точки), и одна клетка заражена на 3 фагов. Также наблюдаемых некоторые неадсорбированный фагов. (C) при Т = 80 мин, две клетки инфицированы одним фаги каждый ушел в литический путь, как показываютг внутриклеточной производства новых фагов (зеленый). Ячейка инфицированных на 3 фагов ушла в лизогенных путь, как указано в производстве mCherry от PRE промотора (красный). (D) при Т = 2 ч, литический путь привел к лизису клетки (клетки взорвалась), а лизогенных клетка разделена §.
§ левой панели Рисунок 7 (C) и (D) перепечатаны с сотового, 141, Ланьин Цзэн, Сэмюэл О. Скиннер, Chenghang Zong, Жан Сиппи, Майкл Feiss, и Идо Голдинг, принятие решений на субклеточном уровне определяет исход инфекции бактериофаг, 682-691, авторские права (2010), с разрешения Elsevier.
| Процедить имя | Соответствующие генотипом | Источник / ссылка |
| Бактериальные штаммы | ||
| LE392 | вирФа | Джон Cronan, Университет штата Иллинойс |
| Фаг штаммов | ||
| λ LZ1 | GPD-EYFP, cI857 SAM7 D-eyfp B :: kanR | Цзэн и др. 5. |
| λ LZ2 | GPD-мозаику, одинаковый генотип, как λ LZ1 | Цзэн и др. 5. |
| Плазмиды | ||
| рр RE - mCherry | mCherry под контролем P RE, усилитель R | Цзэн и др. 5. |
| pPLate * D | GPD под контролем λ поздний промотор, усилитель R | Цзэн и др. 5. |
Таблица 1. Бактериальных штаммов,фаги и плазмиды, используемые в этой работе.
| Плотность ρ (г / мл) | CsCl (г) | SM (мл) | Показатель преломления η |
| 1,30 | 39 | 86 | 1,3625 |
| 1,50 | 67 | 82 | 1,3815 |
| 1,70 | 95 | 75 | 1,3990 |
Таблица 3. CsCl растворов, приготовленных в SM буфера (100 мл) для шага градиента.
Discussion
Штаммы бактерий, фагов и плазмид:
Штамма LE392 является supF. Он был выбран для подавления SAM7 мутаций в геноме фага (см. Таблицу 1 для подробностей). Таким образом, индуцированные лизогенов в конечном итоге лизиса и отпустите частиц фага, так же как инфицированные клетки, которые выбрали литического пути. Лизогенных клетках, выращенных при 30 ° C в связи с наличием чувствительных к температуре CI 857 аллелей в геноме фага. После тепловой индукции, GPD-EYFP и дикого типа GPD будут совместно выразили из генома λ LZ1 и плазмиды pPlate * D соответственно. В результате капсида вновь созданного фага λ LZ2 содержит смесь GPD-EYFP и ВВП белков. Эта мозаика фага является грубой и достаточно флуоресцентные, чтобы обнаружить отдельные фаги 5. рр RE - mCherry, корреспондент плазмиды используются для обнаружения выбор лизогенных pathwaу. RE промотора P активируется CII при установлении лизогении 1,11. рр RE - mCherry 5 была получена из ПЭ-GFP 11 по замене GFP с mCherry 12. Для получения дополнительной информации см. наши предыдущие работы 5.
Параметры роста Состояние:
Во время индукции lysogen (раздел 1), легкая тряска при 180 оборотов в минуту дает хороший выход вируса 13. Использование глюкозы в питательной среде следует избегать, поскольку метаболизм глюкозы создает кислую продуктов обмена веществ, и зрелых частиц лямбда неустойчивы при кислом рН 13. Добавление MgSO 4, направленные на стабилизацию капсида фага 3. Для проведения фагов дикого типа CI (CI вместо 857), lysogen может быть вызвано использованием ДНК-повреждающих агентов митомицин C 3. В шаге 1.3, инкубации при 37 ° C обычно не должна превышать 90 минут. Это USEF ул проверить плотность клеток на OD 600 каждые 30 мин. Для хорошего лизат, OD 600 падает до 0,2 или меньше, а оставшиеся OD 600 является результатом клеточного мусора. Инкубация слишком долго может привести к снижению доходности фагов, так как вновь созданные фага может начать вводить свою ДНК в клетку мусора. Для получения видимого диапазона фага (по крайней мере, 1 х 10 11 частиц фага) с шагом 2,11 и 2,13, расти по меньшей мере 500 мл культуры в шаге 1.2. Кроме того 0,2% мальтозы в ростовой среде в шагах 5.1 и 5.2 направлена на индукции выражение из баранины, рецептор для адсорбции фага лямбда 3,14. 1000-кратного разведения вместо 100-кратного в шаге 5,2 направлена на снижение уровня mCherry фон от репортера плазмиды RE PP - mCherry. В шаге 5.5 для инъекции ДНК фага запуска, 35 ° C выбрана, чтобы избежать индукции чувствительные к температуре cI857 аллеля.
Фаг Очистка:
jove_content "> фага стадии очистки (шаги 2.1 через 2,11) может быть заменен другими протоколами очистки 5, однако окончательное ультрацентрифугирования через CsCl равновесный градиент (Шаги 2.12 и 2.13) неизбежна. Бакет-роторы необходимые шаги в 2,10 и 2,12 до обеспечить резкое видимых полос фага. Получение чистой складе фага может легко занять до недели, так что необходимо проверить титр фага по пути, чтобы убедиться, что все идет не так во время промежуточных шагов.Фаг-разгрузочные работы:
Во время всех процедур очистки в разделе 2, очень важно для обработки фага лизата осторожно, чтобы избежать сдвига фага хвосты из фага головы. В ячейке инфекции в разделе 5 (например, шаги 5.5 через 5,7), важно также, чтобы избежать сдвига фаговых частиц из инфицированных клеток. Заметим, что если фаг стриженый от инфицированной клетки после введения его ДНК, в результате "темных" инфекцию, то есть винфекцией результат будет наблюдаться в эксперименте, но заражение фагом не будет. Чтобы свести к минимуму такие проблемы, мы используем широкий наконечник пипетки при обработке фагов или фаг / смеси клеток.
DAPI тестирования:
Окрашивание фага акции с DAPI (раздел 4) представляет собой быстрый и эффективный метод, чтобы проверить чистоту фага акций. Она также может быть использована для проверки возможного ухудшения существующего фонда фага с течением времени. Для чистого акции, ко-локализацию YFP и DAPI сигналов под флуоресцентным микроскопом должна быть близка к 100%. Как правило, мы видим, что менее 1% из мест YFP не содержат DAPI (представляющих капсид без вирусного генома), который указывает, что эти частицы не удалось успешно упаковать вирусной ДНК или уже вводят свою ДНК в другом месте. Менее 1% пятен DAPI не содержат YFP (соответствующий нефлуоресцентного фаги). Если это не так, то Шаги 2,12 через 2,14 необходимости повторять в оrder, чтобы очистить еще раз. Что касается визуализации параметров, микроскоп установки в шаге 4.3, не так критично, как в разделе 5, потому что нет долгосрочных живых клеток изображений здесь не требуется. Однако, сохранив те же параметры микроскопа, как и в разделе 5 полезна, если кто-то хочет, чтобы откалибровать интенсивности флуоресценции одной частицы фага. Если PBS-агарозы плиты не очень чистые, или слишком много DAPI краситель используется, некоторые DAPI пятна, соответствующие ДНК фага может быть окружен с "гало". Если слишком мало DAPI краситель используется, сигнал из канала DAPI может быть очень слабым.
Микроскоп система:
Для изображений в разделе 6, мы используем коммерческий инвертированный микроскоп эпифлуоресцентной (Eclipse TE2000-E, Nikon) с целью 100x (план Fluo, числовая апертура 1,40, масло погружение) и стандартный набор фильтров (Nikon). Флуоресценции источником света является дуговая лампа с контролем интенсивности света. Следующие функции компьютерного управления: X, Y и Z роsition; светлого поля и флуоресценции ставни, и флуоресцентный фильтр выбора. Автофокус функция не требуется. В противном случае фокус может легко отойти в замедленной фильма (обычно 4 часа). Возможность приобрести несколько (х, у) позиции в каждый момент времени является полезной, поскольку она позволяет проследить несколько событий инфекции в параллель. Мы обычно приобретают 8 финала позиции в каждом фильме, следуя до 100 событий инфекции. Камеры мы используем охлаждением ПЗС-матрицей с 512х512 16х16 мкм камера с динамическим диапазоном 16 бит (Cascade512, Фотометрия). Приобретение осуществляется с помощью программного обеспечения MetaMorph (Molecular Devices). Микроскоп должен быть помещен в контролируемой температурой комнаты, в качестве альтернативы, микроскоп должен быть окружен с контролируемой температурой камеры.
Image Acquisition:
Для живых клеток изображениями, очень важно, чтобы избежать ненужного воздействия на образец, который может привести к обесцвечиванию и фоtotoxicity. Поэтому лучше сначала охарактеризовать вашу систему, чтобы найти оптимальную освещенность, которая позволяет флуоресцентной детекции хотя и не приводит к чрезмерному обесцвечивания или ингибирования роста клеток. Для получения хорошего изображения флуоресценции, играть с интенсивности возбуждающего света, время экспозиции камеры и усиления. В шага 6,2-6,3, 10 мин интервал кадров выбрано с целью минимизации воздействия света. В каждом кадре, только один в фокусе изображения необходимо в фазово-контрастный (для сотовых признания) и флуоресцентных каналов (для определения судьбы клеток). В первый момент времени, однако, несколько Z-положение изображения по каналу YFP требуется, чтобы захватить все заражении фагов на клеточной поверхности. YFP время экспозиции в начальный кадр может также должны быть выше, чем для покадровой фильма в более поздние сроки.
Анализ изображений:
Очень тщательно рассчитывать фагов частиц вокруг клеточной поверхности в шаге 7.1. Какотмечалось выше, мы принимаем ряд Z-стеки YFP через канал в шаге 6.2. Однако, это все еще может оставить некоторые флуоресцентные частицы фага вне фокуса, которая бросает вызов счета. Длина ячейки в начальной сроки измеряются с помощью программного обеспечения Metamorph. Длина ячейки также может быть измерена ImageJ или других программных средств. Кроме того, автоматизированный дом, построенный Matlab программа может быть очень полезным в получении информации, такой как флуоресценции со временем меняются вдоль клеточных клонов.
Disclosures
Нет конфликта интересов объявлены.
Acknowledgments
Мы благодарны Майклу Feiss и Жан Сиппи для руководства по созданию фагов и очистки. Мы благодарим Майкла Elowitz за предоставление программного обеспечения клеточного распознавания, Schnitzcell. Работа в Голдинга лаборатории при поддержке грантов от Национального института здоровья (R01GM082837), Национального научного фонда (082265, PFC: Центр по физике живых клеток), Welch фонда (грант Q-1759) и гуманитарных наук Пограничного Программа (RGY 70/2008).
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma-Aldrich | D4527-10KU | |
| RNase | Sigma-Aldrich | R4642-10MG | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| SM buffer | TEKnova, Inc. | S0249 | |
| NZYM | TEKnova, Inc. | N2062 | |
| CsCl | Sigma-Aldrich | C3011-250G | |
| Syringe | BD Biosciences | 309585 | |
| Needle | BD Biosciences | 305176 | |
| Dialysis cassette | Thermo Fisher Scientific, Inc. | 66333 | |
| Microscope slide | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti ultra-clear tube | Beckman Coulter Inc. | 344060 | |
| SW60Ti ultra-clear tube | Beckman Coulter Inc. | 344062 | |
| SW40Ti rotor | Beckman Coulter Inc. | 331302 | |
| SW60Ti rotor | Beckman Coulter Inc. | 335649 | |
| Refractometer | Fisher Scientific | 13-947 | |
| Epifluorescence microscope | Nikon Instruments | Eclipse TE2000-E | |
| Table 2. Reagents and equipment. | |||
References
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S.
- Ptashne, M. A genetic switch : phage lambda revisited. , 3rd edn, Cold Spring Harbor Laboratory Press. (2004).
- Hendrix, R. W. Lambda II. , Cold Spring Harbor Laboratory. (1983).
- Hershey, A. D. The Bacteriophage lambda. , Cold Spring Harbor Laboratory. (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. Current protocols in molecular biology. , John Wiley & Sons. (1994).
- Sambrook, J., Russell, D. W. Molecular cloning : a laboratory manual. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Fasman, G. D. Practical handbook of biochemistry and molecular biology. , CRC press. (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Personal communication with M. Feiss. , Forthcoming.
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).

{kind=link}