

Summary
Farmacoterapia sostituzione della dopamina con L-DOPA è il trattamento più comunemente utilizzato sintomatico della malattia di Parkinson, ma è accompagnata da effetti collaterali tra cui i movimenti involontari anomali, discinesia definito
Abstract
MALDI Imaging spettrometria di massa (IMS) è un approccio potente che facilita l'analisi spaziale delle specie molecolari in campioni biologici di tessuto 2 (Fig.1). Una sezione di tessuto 12 um sottile è coperto con una matrice MALDI, che facilita desorbimento e ionizzazione di peptidi e proteine intatte che possono essere rilevati con un analizzatore di massa, in genere utilizzando un MALDI TOF / spettrometro di massa TOF. Generalmente centinaia di picchi può essere valutata in una singola sezione di tessuto cerebrale di ratto. In contrasto con tecniche di imaging comunemente utilizzate, questo approccio non richiede la conoscenza preventiva delle molecole di interesse e consente un'analisi incontrollato e completa di più specie molecolari mantenendo elevata specificità e sensibilità molecolare 2. Qui si descrive un approccio basato MALDI IMS per la comprensione della regione specifici profili di distribuzione di neuropeptidi nel cervello di ratto di un animale modello di malattia di Parkinson (PD).
PD è una comune malattia neurodegenerativa con una prevalenza di 1% per le persone sopra i 65 anni di età 3,4. Il trattamento più comune sintomatica si basa sulla sostituzione della dopamina con L-DOPA 5. Tuttavia questa è accompagnata da gravi effetti collaterali tra cui i movimenti involontari anomali, denominati L-DOPA indotta discinesie (LID) 1,3,6. Uno dei più importanti cambiamento molecolare in LID è una sovraregolazione del mRNA oppioide prodynorphin precursore 7. I peptidi dinorfina modulano la neurotrasmissione in aree cerebrali che sono essenzialmente coinvolte nel controllo del movimento 7,8. Tuttavia, ad oggi i peptidi oppioidi esatti che provengono dalla trasformazione del precursore neuropeptide non sono state caratterizzate. Pertanto, abbiamo utilizzato MALDI IMS in un modello sperimentale animale di malattia di Parkinson e di L-DOPA discinesia indotta.
Spettrometria di massa MALDI immagini dimostrata particolarmente vantaggiosa rispetto a neuropeptide caratterizzazionezione, dal momento che gli approcci comunemente usati basati su anticorpi si rivolge sequenze peptidiche noti e precedentemente osservate modificazioni post-traduzionali. Al contrario MALDI IMS possono svelare nuovi prodotti per la lavorazione del peptide e quindi rivelare nuovi meccanismi molecolari di modulazione neuropeptide di trasmissione neuronale. Mentre la quantità assoluta di neuropeptidi non può essere determinata mediante MALDI IMS, l'abbondanza relativa degli ioni peptidici possono essere delineate dagli spettri di massa, dando intuizioni circa la modifica dei livelli di salute e malattia. Negli esempi qui presentati, l'intensità dei picchi di dinorfina B, alpha-neoendorphin e la sostanza P sono risultati significativamente aumentati nel dorsolaterale, ma non il dorsomediale, striato di animali con discinesia grave che interessano più del viso, muscoli del tronco e orolingual (Fig. 5). Inoltre, MALDI IMS ha rivelato una correlazione tra la gravità della discinesia e dei livelli di des-tirosina alfa-neoendorphin, che rappresenta un meccanismo finora sconosciuto di inattivazione funzionalezione di dinorfine nel corpo striato, come la rimozione di N-terminale tirosina riduce la dinorfina del recettore oppioide capacità legante 9. Questo è il primo studio sulla caratterizzazione neuropeptide in LID usando MALDI IMS ed i risultati evidenziano il potenziale della tecnica per l'applicazione in tutti i campi della ricerca biomedica.
Protocol
Il protocollo viene regolata allo scopo di analisi statistica dei dati IMS MALDI da più sezioni di cervello di ratto, tipicamente 20-30 sezioni, e consiste di cinque differenti fasi comprendenti preparazione del tessuto, matrice applicazione, MALDI-TOF MS, valutazione dei dati, e neuropeptide identificazione. Le procedure sono descritte e descritti in dettaglio più sotto:
1. Tissue preparazione
Questo procedimento comprende la raccolta dei campioni tissutali rispettivi come pure il tessuto sezionamento per l'analisi IMS. Un obiettivo particolare di proteine e analisi peptide è quello di evitare la degradazione proteolitica. Perciò è essenziale lavorare velocemente e diligente durante la dissezione dei tessuti.
- Sacrifica ratti (tipicamente 250-300 g) per decapitazione, rimuovere cervello di ratto entro un massimo post-mortem tempo di 30s <e al congelamento in polvere di ghiaccio secco prima del trasferimento a -80 ° C freezer. Più veloce di congelamento con azoto liquidoaumentare il rischio di microtears nei tessuti cerebrali, che influenzerà negativamente la cristallizzazione della matrice e quindi ridurre la qualità MS (Fig. 2D). Cervelli intere possono essere conservati per diversi anni prima di sezionare senza perdita di qualità del segnale MS.
- Tagliare il tessuto congelato con un microtomo criostato a 12 micron e disgelo fette di montaggio su sezioni di tessuto conduttivo vetrini MALDI (ossido di stagno indio vetrini rivestiti, Bruker Daltonics) o MALDI bersaglio (Fig. 2A-C).
- Sezioni a secco per 15 minuti sotto vuoto e conservare i vetrini a -80 ° C fino ad ulteriore utilizzo. Le sezioni di tessuto devono essere analizzate nel più breve tempo possibile dopo sezionamento, anche se conservato a -80 ° C. Troviamo che la qualità del segnale MS sarà notevolmente ridotta dopo un anno di stoccaggio. Al fine di ridurre l'ossidazione delle proteine e peptidi, l'aria nel serbatoio può essere sostituito con un gas inerte (ad esempio argon o azoto).
2. Matrix applicazione
La matrice domandastep ha un impatto significativo sulla qualità spettro e richiede l'ottimizzazione dei parametri multipli a seconda del tipo di tessuto, nonché l'analita di interesse. Questi fattori includono parametri chimici, quali il tipo di matrice, matrice di concentrazione, pH, lavaggio dei tessuti e modificatori organici così come le impostazioni strumentali tra cui il volume di deposito, risoluzione laterale e numero di deposizioni 10 (Fig. 2D). Per gli esperimenti su larga scala, è di grande importanza per ridurre la varianza, ad esempio applicando la matrice di tutte le sezioni entro un giorno e dallo stesso operatore. Sebbene vi siano molte strategie per applicare la soluzione di matrice come mediante sublimazione oa spruzzo, la deposizione automatizzata di array di goccioline piccola matrice, circa 100-150 picolitri dimensioni, è stato usato con successo per l'analisi di piccole proteine e neuropeptidi in vari tessuti , compresi sezioni cerebrali 9, 10,11, 12, 13.
- Scongelare le sezioni in un essiccatore per 1 hil nostro.
- Assicurarsi che l'esperimento è accecata da una persona diversa dal gestore. Re-label tutti i campioni.
- Lavare le sezioni 1x in 70% etanolo (EtOH, a temperatura ambiente, RT) per 10 sec e due volte in 95% EtOH (RT) per 10 sec. Per gli esperimenti di grandi dimensioni, per eseguire il lavaggio tutti vetrata scorre insieme utilizzando una cuvetta per minimizzare variazione.
- Asciugare le sezioni in un essiccatore per 10 minuti.
- Valutare le sezioni di tessuto al microscopio e verificare la distorsione dei tessuti, microtears e piccole crepe che mettere in pericolo la qualità MALDI MS (Fig. 2D).
- Preparare la soluzione fresca matrice costituita da 50 mg / mL DHB in metanolo al 50%, 10% acetato di ammonio 150 mM (Amac) e 0,3% acido trifluoroacetico (TFA) in acqua.
- Matrix applicazione viene effettuata mediante deposizione gocciolina discreto in un modello rettangolare con una stampante a getto d'inchiostro chimico (CHIP, Shimadzu). Il primo passo è di ottimizzare i parametri sperimentali di applicazione matrice di neuropeptide analisi including il numero di goccioline per passaggio, il numero di passaggi. Questo esperimento viene eseguita mediante l'applicazione di array a matrice multipli con parametri applicativi diversi sulla stessa sezione di tessuto, pur facendo in modo che ogni matrice copre le regioni del cervello simili come corpo calloso, corteccia, e nello striato. Lo stesso esperimento deve essere eseguito ogni volta che viene cambiata parametri, comprese le strutture cerebrali differenti, matrici differenti targeting analiti specifici e se solventi matrice differenti sono necessari per l'estrazione di analiti specifici.
- Eseguire la scansione del titolare del vetrino con la sezione di tessuto e allineare il titolare. Definire la matrice per l'applicazione della matrice sulla sezione di tessuto e specificare il luogo cioè la risoluzione spaziale per individuare a distanza. Applicare matrice utilizzando il protocollo ottimizzato sulla stampante a getto d'inchiostro chimico. Per questo esperimento abbiamo usato un protocollo ottimizzato per l'imaging peptide con i parametri di stampa: 10 gocce (100 pl / drop), 10 applicazione passa e un posto per individuare distance di 300 micron.
- Scansione finale matrice avvistato sezioni e salvare l'immagine per la registrazione prima dell'acquisizione MALDI dati (fase 3.4).
- Conservare le sezioni fino ad ulteriore utilizzo in un essiccatore sotto vuoto.
3. MALDI MS acquisizione e l'elaborazione dei dati
L'analisi MS di neuropeptidi viene eseguita su un tempo MALDI strumento di volo (Ultraflex II, Bruker Daltonics, Germania) operano in modalità di riflettore, utilizzando software di acquisizione dati assistita da ogni singolo punto matrice 14. Quindi precisa l'insegnamento dello spazio è un imperativo. È essenziale che la MALDI ottimizzazione, acquisizione ed in particolare gli esperimenti di registrazione target vengono eseguiti dallo stesso operatore che dovrebbero preferibilmente essere a conoscenza dei gruppi sperimentali. In un esperimento su larga scala con vetrini multipli, gli esperimenti MALDI può essere eseguito da un operatore mentre un'altra persona è in funzione della stampante a getto d'inchiostro chimico.
- Caricare vetrini nello spettrometro di massa.
- Controllare la taratura del metodo di acquisizione MALDI utilizzando un basso peso molecolare miscela di taratura del peso (Bruker Daltonics).
- Ottimizzare i parametri di acquisizione.
- Al fine di ottimizzare il segnale MS e per evitare l'ablazione matrice da depositi matrice vicine, la dimensione del laser e il fuoco ottimale sul tessuto deve essere determinato.
- L'energia laser è impostata per garantire la massima qualità da MS come depositi matrice possibili senza elevare la linea di base, riducendo la risoluzione di picco o saturare il rivelatore.
- Valutare il numero massimo di scatti per punto fino a matrice unico rumore viene rilevato, spesso 1000-2000 colpi. Stimare il numero di scatti che dovrebbero essere accumulate e il numero di scatti maturati prima della posizione del laser all'interno di uno spot dovrebbe cambiare. Al fine di campionare ogni punto della matrice in modo uniforme, si accumulano 600 scatti in 25 passi colpo, per un totale di 24 passaggi con un ranmodello di libertà di movimento, da ogni deposizione di matrice.
- Registrare la scansione di tutte le sezioni macchiati in coordinate motore della fase MALDI utilizzando il FlexImaging (v.2.0) 10 ed eseguire l'acquisizione dei dati in modalità batch dal software AutoXexuteBatchRunner.exe.
- Elaborare ogni singolo spettri mediante sottrazione di base (Convex scafo V3), levigante e calibrazione esterno (opzionale), seguita da esportare in un file ASCII (*. Dat, *. Txt o *. In formato csv) 15.
4. Valutazione dei dati
Valutazione finale dei dati comprende messaggio trattamento dei dati e riduzione dei dati da fuoco solo su informazioni di picco, seguito da analisi statistica.
- Come primo passo, le sezioni IMS MALDI sono stati valutati per gli effetti overnormalization. Questo può essere facilmente ottenuto con l'impiego di strumenti di visualizzazione dei dati come FlexImaging (Bruker Daltonics) o BioMap (Novartis). Come primo passo le immagini ioni totali sono evaluated prima di corrente totale di ioni (TIC) normalizzazione, seguita da ispezione manuale di singole immagini di distribuzione di ioni di vari picchi peptide importanti. Se vuoi per le distribuzioni caratteristiche di intensità di picco e se sono legati a caratteristiche dei tessuti (i danni), spotting effetti di qualità o normalizzazione (Fig. 3).
- Delineare regioni di interesse (ad esempio, il corpo striato) in base alle caratteristiche istologiche ed esportare gli spettri corrispondente in formato ascii. Preferibilmente, la normalizzazione di spettri alla corrente ionica totale (TIC) può essere effettuato in questa fase.
- Importare file ASCII in un software di gestione dei dati come l'origine (V.8.1, OriginLab), MATLAB (The MathWorks, Natick, MA, USA) o R 16. Rilevamento di picco può essere eseguita utilizzando strumenti di ricerca di punta inclusi nel software, ad esempio "l'analisi di picco" in origine "o" mspeaks in Matlab. Esportare i peaklists da tutti gli spettri, come un unico file di testo delimitato da tabulazioni.
- Al fine di determinare i confini per bin peptide rilevatopicchi, l'analisi binning viene eseguita utilizzando strumenti software adeguati (ad esempio pbin 17) o in-house scritta script per MATLAB o R. Ecco il file di testo unico contenente tutte le picco raccolto i dati vengono caricati nel software ed i parametri per la determinazione di frontiera di picco sono specificati ad esempio come spesso un picco dovrebbe essere presente in spettri per essere rilevante per l'esperimento. Ad esempio, l'esperimento contiene 2 gruppi di animali, 5 animali in ciascun gruppo, e 100 spettri sono raccolti da ciascun animale e regione di interesse. Si supponga un picco è potenzialmente interessante se è presente almeno nella maggior parte degli animali in un gruppo (3/5) e in almeno metà degli spettri di tali animali (3x50 = 150 spettri), questo darà una percentuale totale del 15% per gli spettri 150 positivi su un totale di 1000 (2x5x100) spettri. Con lo strumento pbin, questa fase produce un singolo file binrange contenente tutte le larghezze scomparto determinati dai dati acquisiti. Al fine di verificare che i confini binsono adeguate, è facile visualizzare i bidoni in origine, unitamente con le tracce originali spettri.
- Peak-area di integrazione può ridurre varianza che è importante per l'analisi statistica. Usiamo un in-house copione scritto per R per calcolare l'area sotto la curva tra i confini di picco determinati al punto 4. Integrata delle aree dei picchi vengono importati in MS Excel (v.2007) e l'analisi statistica mediante test non parametrico spaiato utilizzando lo strumento SAM viene eseguita 18.
5. Peptide di identificazione
Verifica della sequenza delle identità peptide osservate è essenziale per concludere importanza biologica. L'approccio più accurato includono vera top-down determinazione direttamente dal tessuto con la frammentazione peptide mediante spettrometria di massa tandem (MS / MS), ma le concentrazioni di peptide sono richieste elevate per questo tipo di analisi 12,13. Per peptidi a basso abbondanti o peptidi multipli con closem / valori di z (± 0,5%), a-tessuto analisi è compromessa e analisi velina utilizzando una strategia peptidomic viene utilizzata che comprende l'estrazione, separazione e l'identificazione basata su MS di neuropeptidi endogeni. Per l'esperimento qui presentata, il punto centrale era rilevamento peptide oppiaceo, che è una sfida particolare poiché questi peptidi sono piuttosto basse abbondanti neuropeptidi rispetto ad altri spettri. Inoltre, questi peptidi sono piuttosto polare che li rende relativamente idrofilo e difficili da mantenere con estrazione peptide comune e tecniche di separazione .. Quindi abbiamo applicato un protocollo riportato in precedenza per l'estrazione dei tessuti e prefrazionamento peptide oppiaceo in combinazione con LC-MS/MS standard basati identificazione peptide 9,19.
- Raccogliere sezioni coronali di strutture bersaglio di interesse (nucleo accumbens, NAc, caudato putamen, CPU). Montare cervello congelato di ratto in un microtomo criostato e rimuovere il materiale cerebrale circostante (cortex, setto, corpo calloso), con un bisturi. Raccogliere le sezioni (30 micron, n = 50) del sezionato NAc e la CPU e disgelo montare il NAc e le parti della CPU delle sezioni su vetrini differenti.
- Estrarre peptidi off tessuto aggiungendo 100 microlitri TFA 5% ACN/0.1%, incubare per due minuti e raccogliere in provette eppendorf proteine a basso vincolanti. Ripetere questa operazione due volte.
- Eseguire prefrazionamento peptide mediante cromatografia a scambio cationico forte utilizzando stepwise (n = 4) eluizione a maggiore forza ionica 19. Essiccare le campioni sotto vuoto usando un concentratore SpeedVac.
- Analizzare le frazioni peptidiche mediante cromatografia a fase inversa C18 nanoflussi liquida (1100, Agilent Technologies, Santa Clara, CA) interfacciati a elettrospray spettrometria di massa tandem (LC-MS/MS). MS Gli esperimenti sono stati effettuati su un ibrido lineare iontrap / Fourier transform ion ciclotrone risonanza (FTICR) strumento (LTQ FT 7T, Thermo Scientific, Waltham, MA,) Peptide fulscan spettri (m / z 150-2000) Sono stati acquisiti con l'analizzatore FTICR ad alta risoluzione di massa (100 mila), seguita da successiva frammentazione dei 5 picchi di peptidi più intensi del iontrap mediante dissociazione indotta da collisione (CID) 9.
- Identificazione sequenza peptidica è eseguita da corrispondenza database e possono essere integrati da de-novo analisi di sequenziamento. Per la ricerca del database, motori di ricerca disponibili in commercio (Mascot, XTandem o Proteina Prospector) sono impiegati 20. Le ricerche vengono in genere eseguite nei database contenenti sequenze di neuropeptidi noti o prevedibili e le sequenze delle proteine precursori neuropeptidi 21.
6. Risultati rappresentativi
Spettrometria di massa MALDI immagini di sezioni di tessuto striatali come preparato secondo il protocollo descritto qui portato alla rilevazione di più di 1000 picchi corrispondenti a circa 300 specie molecolari (monoisotopico spettri media mostratiin fig. 1). Visualizzazione dei dati per importanti picchi di ioni molecolari è stato realizzato utilizzando il software Imaging Flex e ha mostrato caratteristiche distribuzioni di intensità di picco che sono perfettamente in linea con le caratteristiche anatomiche (Fig.3). Un'ulteriore caratteristica di MALDI IMS è la sua riproducibilità relativamente buono. In questo esperimento, il coefficiente complessivo di varianza per l'intensità dei picchi di tutte le specie rilevate molecolare era del 30%, ma molti picchi visualizzato variazione molto bassa e alta riproducibilità entro gruppi di trattamento (Fig. 4). I relativi dati di picco di intensità di quattro diverse regioni di interesse, compresa la parte dorsolaterale e dorsomediale di striata sia lesionato e intatto sono stati sottoposti ad analisi statistica. Al fine di regolare per confronti multipli simultaneamente, l'analisi statistica è stata effettuata mediante test non parametrico utilizzando lo strumento SAM 18. I cambiamenti più importanti sono stati trovati nella parte dorsolaterale della dopamina-denervato, striato parkinsoniano. Qui SI cambiamenti nei gnificant tra i diversi gruppi di trattamento sono stati osservati per due peptidi dinorfina, dinorfina B e alfa-neoendorphin (Fig.5). In dettaglio, un aumento relativo di due picchi di intensità dinorfina del 50-60% è stata osservata in alto rispetto al animali discinetiche basse animali discinetiche e controlli lesione (p <0,05, F (2, 15) = 12,8 DynB, F = 5,7 ANEO; Fig. 5).

. Figura 1 media MS tracce ottenute da due regioni strettamente connesse dello striato; caudato putamen (CPU) e il nucleo accumbens (NAc). Le due regioni visualizzare diversi profili di MS con alcune specie molecolari in modo univoco espressi in una regione, o ai diversi livelli di intensità di picco (insert, m / z 2028). Il modello di distribuzione spaziale di ogni picco può essere visualizzato usando il software specializzato di imaging (pannello inferiore).
2.jpg "/>
Figura 2 (A) Il cervello è montato su un mandrino criostato utilizzando un mezzo di incorporamento (OTC; freccia)., Si ha cura che l'OTC non contaminare l'area del cervello da sezionare poiché la soppressione ionica causa OTC di peptidi. (B, C) sezioni sottili (≈ 12 um di spessore) sono montati su scongelamento vetrini MALDI compatibili ed essiccato per alcuni secondi per evitare danni congelamento come visto in C. (D) Microtears possono essere difficili da rilevare a occhio nudo , ma mettere in pericolo MALDI cristallizzazione della matrice e cancellare il segnale MALDI MS. La stessa sezione colorate con violetto cresolo rivela microtears e crepe microfotografia (in basso a destra).

Figura 3. Il primo passo nella valutazione dei dati è quello di visualizzare vari picchi differenti su tutta la gamma di massa analizzato (AI). Qui, le sezioni striatali da 9 topi sono stati ripresi con MALDI MS. Visualizzazione del totale i mediasulle attuali rivelerà zone di cospicui intensità di ioni ad alta o bassa (frecce). Queste aree possono essere influenzati da sovra o sotto-normalizzazione effetti e falsare l'analisi dei dati compromettere i risultati. Scarsa definizione anatomica delle distribuzioni di punta rivelano sezioni con picchi generalmente basso rapporto segnale-rumore, ad esempio, le sezioni 3 e 9, con picchi di F I.

Figura 4. Riproducibilità MS tra i gruppi di trattamento può essere valutata calcolando la media MS traccia e l'errore standard per ogni m / z valore (inserti, m / z 722 e 1749). Buona riproducibilità assicura valida analisi statistica.
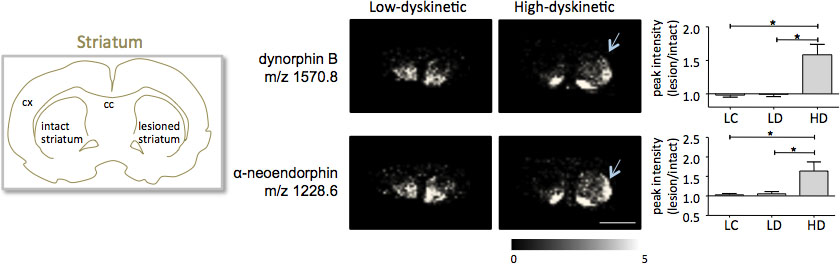
Figura 5. Dinorfina B e alfa-neoendorphin potenze di picco sono significativamente aumentati nel 6-OHDA-Leso, parkinsoniano, striato di alta discinetici animali (HD; frecce) rispetto al gruppo di controllo a bassa discinetica (LD) e della lesione (LC). Intensità dei picchi peptide espresso in termini di media-fold cambio di lato sano ± SEM (lesione / intatta lato). * P <0,05; cx corteccia; cc corpo calloso. Scale bar 5 mm.
Discussion
Ci sono diversi vantaggi di spettrometria di massa MALDI impiegare immagini nello studio di neuropeptidi. L'analisi dei dati imparziale MS può rivelare che nuclei cervello solo specifico, o come nei risultati qui presentati in cui solo la parte dorsolaterale dello striato è associato ad una certa condizione fisiopatologica. Mantenendo le informazioni spaziali è poi possibile ridefinire regioni di interesse per effettuare analisi statistiche con maggiore sensibilità e minore variabilità rispetto all'analisi di sezioni cerebrali intere o con tradizionali studi peptidomics su estratti peptide. Inoltre, è importante realizzare MALDI IMS prontamente in grado di rilevare precedentemente sconosciuti modifiche post-traduzionali, ma analisi strutturali deve seguire per determinare le posizioni esatte amminoacidiche che vengono modificati.
Errori più frequenti nella visualizzazione dei dati IMS MALDI includono la mappatura l'intensità massima di picco di una scala lineare ottica da black (0%) di colore (100%) per ogni sezione della serie sperimentale (figura 3), invece di mappatura tutte le sezioni di una scala comune assoluto dove 100% è l'intensità del picco massimo di tutte le sezioni (figura 5) . Quest'ultimo metodo consente il confronto dei dati di gruppo e la visualizzazione delle differenze tra i gruppi di trattamento.
Uno dei principali ostacoli in analisi MALDI IMS è l'assegnazione di peptidi a picchi di massa specifici. On-tessuto-spettrometria di massa tandem a volte è possibile, ma risulta spesso molto difficile 13,14. Abbiamo trovato che un approccio più tradizionale comprendente un frazionamento preparativa su forte cromatografia a scambio cationico, seguito da LC-MS/MS fase inversa può essere utilizzato con successo per neuropeptidi sequenza molti peptidi e soprattutto oppioidi. Non è ancora raro ottenere una buona qualità di MS / MS che non corrispondono a tutte le voci del database che utilizzano i motori di ricerca comuni come mascotte. In questi casi de novo sequencing-mano è il only opzione. L'ultima prova di identità di picco può essere ottenuta tramite MALDI IMS di sezioni di tessuto dal topo knockout appropriato, ma questo non è sempre disponibile o possibile. Un'alternativa è quella di convalidare i risultati con un metodo diametralmente diverso, ad esempio mediante immunoblotting occidentale o immunoistochimica. Questo può spesso includono alzando gli anticorpi e una notevole quantità di lavoro la convalida dei nuovi anticorpi.
La strategia generale illustrata in questo protocollo è ottimizzato per le grandi dimensioni neuropeptidi MALDI esperimenti IMS tra cui diverse sezioni e delle condizioni sperimentali. Il protocollo è stato ottimizzato in particolare per i peptidi oppioidi e avrà grande impatto in studi futuri, come impiegata in diversi campi di ricerca, compreso il dolore dei meccanismi alla base e la risposta ai farmaci endogena di dipendenza.
Disclosures
Gli autori non hanno nulla da rivelare.
Acknowledgments
Ringraziamo Hanna Warner per aver contribuito i dati per la figura 3 e il Prof. Jonas Bergquist per un contributo prezioso. Il Consiglio svedese della ricerca (Grant 522-2006-6416 (MA), 521-2007-5407 (MA); Il Wiberg Åke della Fondazione (MA, JH), L'Accademia Reale Svedese delle Scienze (MA, JH), e il chimico svedese Society (JH) Si ringraziano per il sostegno finanziario.
References
- Obeso, J. A., Olanow, C. W., Nutt, J. G. Levodopa motor complications in Parkinson's disease. Trends Neurosci. 23, S2-S7 (2000).
- Caprioli, R. M., Farmer, T. B., Gile, J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. MALDI-TOF MS. Anal. Chem. 69, 4751-4760 (1997).
- Obeso, J. A. The evolution and origin of motor complications in Parkinson's disease. Neurology. 55, S13-S20 (2000).
- O, W. H. Noncommunicable Diseases and Mental Health Cluster, Noncommunicable Disease Prevention and Health Promotion Department, Ageing and Life Course. Active Ageing: A Policy framework. , (2002).
- Schapira, A. H. Movement disorders: advances in cause and treatment. Lancet Neurology. , 6-7 (2010).
- Obeso, J. A., Rodriguez-Oroz, M. C., Rodriguez, M., DeLong, M. R., Olanow, C. W. Pathophysiology of levodopa-induced dyskinesias in Parkinson's disease: problems with the current model. Ann. Neurol. 47, S22-S32 (2000).
- Cenci, M. A., Lee, C. S., Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 10, 2694-2706 (1998).
- Andersson, M., Hilbertson, A., Cenci, M. A. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis. 6, 461-474 (1999).
- Hanrieder, J. Alterations of striatal neuropeptides revealed by imaging mass spectrometry. Molecular & Cellular Proteomics. , (2011).
- Cornett, D. S., Reyzer, M. L., Chaurand, P., Caprioli, R. M. MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods. 4, 828-833 (2007).
- Ljungdahl, Imaging Mass Spectrometry Reveals Elevated Nigral Levels of Dynorphin Neuropeptides in L-DOPA-Induced Dyskinesia in Rat Model of Parkinson's Disease. PLoS ONE. 6, e25653 (2011).
- Groseclose, M. R., Andersson, M., Hardesty, W. M., Caprioli, R. M. Identification of proteins directly from tissue: in situ tryptic digestions coupled with imaging mass spectrometry. J. Mass. Spectrom. 42, 254-262 (2007).
- Andersson, M., Groseclose, M. R., Deutch, A. Y., Caprioli, R. M. Imaging mass spectrometry of proteins and peptides: 3D volume reconstruction. Nat. Methods. 5, 101-108 (2008).
- Deininger, S. -O. Imaging Mass Spectrometry. Setou, M. , Springer. Japan. 199-208 (2010).
- Norris, J. L. Processing MALDI Mass Spectra to Improve Mass Spectral Direct Tissue Analysis. Int. J. Mass. Spectrom. 260, 212-221 (2007).
- Ihaka, R., Gentleman, R. R. A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics. 5, 299-314 (1996).
- Mass Spectrometry Binning Software GAB. , Vanderbilt Center for Quantitative Sciences. Nashville, TN. Available from: http://www.vicc.org/biostatistics/software.php (2012).
- Tusher, V. G., Tibshirani, R., Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116-5121 (2001).
- Bergstrom, L., Christensson, I., Folkesson, R., Stenstrom, B., Terenius, L. An ion exchange chromatography and radioimmunoassay procedure for measuring opioid peptides and substance P. Life. Sci. 33, 1613-1619 (1983).
- Falth, M. Neuropeptidomics strategies for specific and sensitive identification of endogenous peptides. Mol. Cell. Proteomics. 6, 1188-1197 (2007).
- Falth, M. SwePep, a database designed for endogenous peptides and mass spectrometry. Mol. Cell. Proteomics. 5, 998-1005 (2006).
