

Summary
Pharmacothérapie remplacement de la dopamine en utilisant la L-DOPA est le traitement le plus couramment utilisé symptomatique de la maladie de Parkinson, mais il est accompagné par des effets secondaires, y compris des mouvements anormaux involontaires, dyskinésie appelé
Abstract
Imagerie MALDI spectrométrie de masse (IMS) est une approche puissante qui facilite l'analyse spatiale des espèces moléculaires dans des échantillons de tissus biologiques 2 (Fig.1). Une coupe de 12 um de tissu mince est recouverte d'une matrice MALDI, ce qui facilite la désorption et l'ionisation des peptides et des protéines intactes qui peuvent être détectés avec un analyseur de masse, typiquement en utilisant une TOF MALDI / spectromètre de masse TOF. Généralement des centaines de pics peuvent être évalués dans une coupe de tissu de cerveau de rat unique. Contrairement aux techniques d'imagerie utilisées couramment, cette approche ne nécessite pas de connaissance préalable des molécules d'intérêt et permet d'analyser non supervisée et complète de plusieurs espèces moléculaires tout en conservant sa spécificité moléculaire et de la sensibilité 2. Nous décrivons ici une approche basée sur IMS MALDI pour élucider les profils de distribution spécifiques à la région de neuropeptides dans le cerveau de rat de la maladie d'un animal modèle de la maladie de Parkinson (PD).
PD est une maladie neurodégénérative fréquente avec une prévalence de 1% pour les personnes de plus de 65 ans de 3,4. Le traitement le plus commun des symptômes est basée sur remplacement de la dopamine en utilisant la L-DOPA 5. Cependant ceci est accompagné par des effets secondaires graves, y compris des mouvements anormaux involontaires, appelés L-DOPA dyskinésies induites (LID) 1,3,6. Un des changements les plus important moléculaire dans LID est une régulation positive de l'ARNm prodynorphine précurseur opioïde 7. Les peptides dynorphine moduler la neurotransmission dans les zones du cerveau qui sont essentiellement impliqués dans le contrôle des mouvements 7,8. Toutefois, à ce jour les peptides opioïdes exactes qui proviennent de la transformation du précurseur neuropeptide n'ont pas été caractérisé. Par conséquent, nous avons utilisé MALDI IMS dans un modèle animal de la maladie de Parkinson expérimentale et la L-DOPA dyskinésie induite.
Imagerie MALDI spectrométrie de masse s'est avérée particulièrement avantageuse par rapport à la caractérisation neuropeptidetion, car les approches d'anticorps couramment utilisés cibles basées sur des séquences peptidiques connues et observées auparavant modifications post-traductionnelles. En revanche MALDI IMS peut démêler nouveaux produits de traitement de peptides et de révéler ainsi de nouveaux mécanismes moléculaires de la modulation de la transmission neuronale neuropeptide. Bien que le montant absolu des neuropeptides ne peut pas être déterminée par MALDI IMS, l'abondance relative des ions peptidiques peuvent être délimitées à partir des spectres de masse, donnant un aperçu sur l'évolution des niveaux de santé et la maladie. Dans les exemples présentés ici, les intensités des pics de la dynorphine B, alpha-neoendorphin et la substance P ont été trouvés à être significativement augmentée dans le dorsolatéral, mais pas le dorsomédial, le striatum des animaux avec des dyskinésies sévères impliquant visage, le tronc et les muscles orolingual (Fig. 5). En outre, MALDI IMS a révélé une corrélation entre la sévérité de la dyskinésie et les niveaux de des-tyrosine alpha-neoendorphin, ce qui représente un mécanisme jusqu'alors inconnu de la fonctionnelle inactivationvation de dynorphines dans le striatum que la suppression de la N-terminal tyrosine réduit des récepteurs opioïdes du dynorphine de la capacité de liaison 9. Il s'agit de la première étude sur la caractérisation neuropeptide dans couvercle à l'aide MALDI IMS et les résultats mettent en évidence le potentiel de la technique pour l'application dans tous les domaines de la recherche biomédicale.
Protocol
Le protocole est ajusté dans le but de l'analyse statistique des données d'IMS MALDI à partir de plusieurs sections de cerveau de rat, typiquement 20-30 sections, et se compose de cinq étapes différentes comprenant la préparation des tissus, matrice d'application, MALDI-TOF MS analyse, l'évaluation des données, et le neuropeptide l'identification. Les procédures sont exposés et décrits de façon plus détaillée ci-dessous:
1. Préparation des tissus
Cette procédure comprend la collecte des échantillons de tissus respectifs ainsi que des tissus de sectionnement pour l'analyse IMS. Un objectif particulier en protéines et en analyse des peptides est d'éviter la dégradation protéolytique. Par conséquent, il est essentiel de travailler vite et avec diligence lors de la dissection des tissus.
- Sacrifiez les rats (typiquement 250-300 g) par décapitation, retirer le cerveau du rat dans un délai maximum post-mortem de temps de 30s et moins de gel sur la glace sèche en poudre avant de les transférer à -80 ° C congélateur. Une congélation plus rapide en utilisant l'azote liquideaugmenter le risque de microfissures dans le tissu cérébral, ce qui affectera négativement la cristallisation matrice et de réduire ainsi la qualité de MS (Fig. 2D). Cerveaux entiers peuvent être stockés pendant plusieurs années avant de sectionner sans perte de qualité du signal MS.
- Couper les tissus congelés sur un microtome cryostat à 12 tranches de um et des coupes de tissus dégel de montage sur lames de verre conductrices MALDI (diapositives d'oxyde d'indium étain recouvertes, Bruker Daltonics) ou MALDI cibles (Fig. 2A-C).
- Sécher les sections pendant 15 min sous vide et conserver les lames à -80 ° C jusqu'à l'utilisation. Les coupes de tissus doivent être analysés dans le délai le plus court possible après la coupe, même s'ils sont conservés à -80 ° C. Nous constatons que la qualité du signal MS sera sensiblement réduit après un an de stockage. Afin de réduire l'oxydation des protéines et des peptides, l'air dans le récipient de stockage peut être remplacé par un gaz inerte (par exemple l'argon ou l'azote).
2. Matrice d'application
L'application la matriceétape a un impact significatif sur la qualité du spectre et nécessite une optimisation de plusieurs paramètres en fonction du type de tissu, ainsi que l'analyte d'intérêt. Ces facteurs comprennent les paramètres chimiques tels que le type de matrice, de la concentration de matrice, le pH, le lavage des tissus et des modificateurs organiques ainsi que les paramètres instrumentaux dont le volume des dépôts, la résolution latérale et le nombre de dépôts 10 (Fig. 2D). Pour les expériences à grande échelle, il est d'une grande importance pour réduire la variance, par exemple en appliquant la matrice de toutes les sections en une journée et par le même opérateur. Bien qu'il existe de nombreuses stratégies pour appliquer la solution matrice telle que par sublimation ou par pulvérisation, le dépôt automatique des tableaux de gouttelettes de la matrice de petites, environ 100-150 picolitres de taille, a été utilisée avec succès pour l'analyse des petites protéines et des neuropeptides dans les différents tissus , dont 9 coupes de cerveau, 10,11, 12, 13.
- Décongelez les sections dans un dessiccateur pendant 1 hnotre.
- Assurez-vous que l'expérience est aveuglé par une personne autre que l'exploitant. Re-étiquettes de tous les échantillons.
- Laver les coupes 1x dans l'éthanol 70% (EtOH, à la température ambiante, RT) pendant 10 secondes et deux fois en 95% EtOH (RT) pendant 10 sec. Pour les expériences grandes, effectuer le lavage de tous les lames de verre ensemble en utilisant une cuvette afin de minimiser la variation.
- Sécher les sections dans un dessiccateur pendant 10 min.
- Évaluer les coupes de tissus au microscope et vérifier la distorsion du tissu, microfissures et de petites fissures qui nuira à la qualité de spectrométrie de masse MALDI (Fig. 2D).
- Préparer une solution fraîche matrice composée de 50 mg / ml dans le méthanol DHB 50%, 10% d'acétate d'ammonium 150 mM (AMAC) et 0,3% d'acide trifluoroacétique (TFA) dans l'eau.
- Matrice d'application est effectuée par le dépôt de gouttelettes discret dans un motif rectangulaire en utilisant une imprimante jet d'encre chimique (CHIP, Shimadzu). La première étape consiste à optimiser les paramètres expérimentaux de l'application de matrice pour l'analyse neuropeptide including le nombre de gouttelettes par passage, nombre de passes. Cette expérience est réalisée en appliquant les tableaux matriciels multiples avec différents paramètres de l'application sur la même coupe de tissu, tout en veillant à ce que chaque tableau est couvrant les régions du cerveau similaires tels que le corps calleux, du cortex, et du striatum. La même expérience doit être effectuée chaque paramètres de temps sont changés, y compris les structures cérébrales différentes, des matrices différentes ciblant analytes spécifiques, et si la matrice solvants différents sont nécessaires pour l'extraction d'analytes spécifiques.
- Scannez le support de diapositives en verre avec la coupe de tissu et d'aligner le titulaire. Définissez votre tableau pour l'application de matrice sur la coupe de tissu et de préciser l'endroit résolution spatiale soit de repérer à distance. Appliquer la matrice en utilisant le protocole optimisé sur l'imprimante jet d'encre chimique. Pour cette expérience, nous avons utilisé un protocole optimisé pour l'imagerie peptide avec les paramètres d'impression suivants: 10 gouttes (100 pl / chute), 10 application passe et un endroit pour repérer distance de 300 um.
- Numérisation finale matrice repéré sections et enregistrer l'image pour l'enregistrement avant MALDI acquisition de données (étape 3.4).
- Conserver les sections jusqu'à l'utilisation dans un dessicateur sous vide.
3. Spectrométrie de masse MALDI d'acquisition de données et le traitement
L'analyse des neuropeptides MS est effectuée sur une période de vol aux instruments MALDI (Ultraflex II, Bruker Daltonics, Allemagne) fonctionnant en mode réflecteur, en utilisant un logiciel d'acquisition de données assistée de tous les points une seule matrice 14. Par conséquent précise l'enseignement spatiale est impératif. Il est essentiel que l'optimisation MALDI, l'acquisition et en particulier les expériences d'enregistrement cibles sont effectuées par le même opérateur qui devraient de préférence être aveuglé les groupes expérimentaux. Dans une expérience à grande échelle avec des lames de verre multiples, les expériences MALDI peut être effectuée par un opérateur tandis qu'une autre personne est d'utiliser l'imprimante à jet d'encre chimique.
- Charger les lames de verre dans le spectromètre de masse.
- Vérifier l'étalonnage de la méthode de l'acquisition MALDI l'aide d'un faible mélange moléculaire étalonnage des poids standard (Bruker Daltonics).
- Optimiser les paramètres d'acquisition.
- Afin d'optimiser le signal EM et pour éviter des dépôts d'ablation matrice matrice voisins, la taille du laser et le point optimale sur le tissu doit être déterminée.
- L'énergie laser est réglé pour assurer la qualité maximale de MS en tant que dépôts de matrice grand nombre possible sans élever le niveau de référence, la réduction de résolution de crête ou de saturation du détecteur.
- Évaluer le nombre maximum de coups par point jusqu'à ce que la matrice de bruit est détecté seulement, souvent 1000-2000 tirs. Estimer le nombre de coups de feu qui devrait être accumulés et le nombre de coups acquis avant la position du laser dans un endroit doit changer. Afin de goûter à chaque point de matrice uniforme, nous accumulons 600 coups en 25 étapes shot, pour un nombre total de 24 étapes en utilisant un ranmotif de liberté de mouvement, de chaque dépôt de matrice.
- Inscrivez-vous l'analyse de toutes les sections repérés en coordonnées moteur de la scène en utilisant le MALDI FlexImaging (v.2.0) 10 et l'acquisition des données en mode batch par le logiciel AutoXexuteBatchRunner.exe.
- Traiter chaque spectres unique par le biais de base de soustraction (Enveloppe convexe V3), le lissage et l'étalonnage externe (en option), suivi par l'exportation sous forme de fichier ascii (*. Dat, *. Txt ou au format *. Csv) 15.
4. L'évaluation des données
Final d'évaluation de données comprend le post-traitement des données et la réduction des données par se concentrer uniquement sur l'information de pointe, suivie d'une analyse statistique.
- Comme première étape, les sections MALDI IMS ont été évaluées pour leurs effets overnormalization. Ceci peut être facilement réalisé en utilisant des outils de visualisation de données tels que FlexImaging (Bruker Daltonics) ou BioMap (Novartis). Dans une première étape les images ioniques totales sont evaluée avant courant ionique total (CIT) de normalisation, suivie d'une inspection manuelle de simples images de la distribution des ions de divers pics peptidiques éminents. Rechercher des caractéristiques de pointe et distributions d'intensité si elles sont liées aux caractéristiques des tissus (dommages), le repérage des effets de qualité ou de normalisation (Fig. 3).
- La délimitation des régions d'intérêt (par exemple, le striatum) en fonction des caractéristiques histologiques et d'exporter les spectres correspondant au format de fichier ascii. De préférence, la normalisation des spectres pour le courant ionique total (CIT) peut être effectuée à ce stade.
- Importer des fichiers ASCII dans un logiciel de traitement des données telles que l'origine (v.8.1, Originlab), MATLAB (The MathWorks, Cambridge, MA, Etats-Unis) ou R 16. Détection des pics peut être effectuée en utilisant des outils de recherche de pointe inclus dans le logiciel, par exemple "l'analyse de pointe" dans l'origine ou "" dans mspeaks Matlab. Exporter les peaklists de tous les spectres en un seul fichier texte unique onglet délimité.
- Afin de déterminer les frontières bin pour le peptide détectépics, l'analyse est effectuée en utilisant binning outils logiciels appropriés (p. ex pBin 17) ou en interne écrit des scripts pour MATLAB ou R. Ici, le seul fichier texte contenant tous les pic repris les données sont chargées dans le logiciel et les paramètres pour la détermination des frontières de pointe sont précisées telles que combien de fois un pic doit être présent dans les spectres afin d'être pertinente pour l'expérience. Par exemple, l'expérience contient 2 groupes d'animaux, 5 animaux dans chaque groupe, et de 100 spectres sont recueillies auprès de chaque animal et de la région d'intérêt. Prendre une crête est potentiellement intéressant si elle est présente dans au moins la majorité des animaux dans un groupe (3/5) et dans au moins la moitié des spectres de ces animaux (3x50 = 150 spectres), ce qui donne une proportion totale de 15% pour les spectres 150 positifs sur le total 1000 (2x5x100) spectres. Utilisation de l'outil pBin, cette étape donne un fichier unique contenant binrange toutes les largeurs bin déterminées à partir des données acquises. Afin de vérifier que les frontières binsont appropriés, il est facile de visualiser les bacs à l'origine en collaboration avec les traces des spectres originaux.
- Pic-zone d'intégration permet de réduire la variance qui est important pour l'analyse statistique. Nous utilisons un script en interne écrite pour R pour calculer l'aire sous la courbe entre les frontières de pointe déterminées à l'étape 4. Intégrés des zones de pointe sont importés dans MS Excel (v.2007) et l'analyse statistique au moyen de tests non paramétriques non apparié en utilisant l'outil SAM est effectuée 18.
5. Identification de peptides
Séquence de vérification des identités peptidiques observés est essentielle en vue de conclure la pertinence biologique. L'approche la plus précise notamment vrai top-down détermination directement à partir de tissu en utilisant la fragmentation peptidique par des moyens de spectrométrie de masse tandem (MS / MS), bien que les concentrations élevées de peptides sont nécessaires pour ce type d'analyse 12,13. Pour des peptides de faible abondance ou de peptides multiples avec le ClosEM / valeurs z (± 0,5%), en analyse de tissus est altérée et l'analyse des tissus en utilisant une stratégie hors peptidomic est utilisé qui comprend l'extraction, la séparation et l'identification basée sur MS de neuropeptides endogènes. Pour l'expérience présentée ici, le point central était sur la détection peptide opioïde, qui est un défi particulier puisque ces peptides sont très faibles abondante par rapport à d'autres neuropeptides dans les spectres. En outre, ces peptides sont plutôt polaire qui les rend relativement hydrophile et difficile à conserver à l'extraction peptide commun et des techniques de séparation .. Par conséquent, nous avons appliqué un protocole indiqué précédemment pour l'extraction des tissus et préfractionnement peptide opioïde en combinaison avec LC-MS/MS normalisées sur la base d'identification peptide 9,19.
- Recueillir des coupes coronales de structures cibles d'intérêt (noyau accumbens, du CNA; caudé putamen, CPU). Montez le cerveau de rat congelé dans un microtome cryostat et enlever de la matière cérébrale environnante (cortex, le septum, le corps calleux) avec un scalpel. Recueillir des sections (30 um, n = 50) de la disséqué NAc et CPU et de dégel monter le CNA et les parties des sections CPU sur lames de verre différents.
- Extrait peptides hors des tissus en ajoutant 100 uL de TFA 5% ACN/0.1%, incuber pendant deux minutes et recueillir dans des tubes eppendorf faible teneur en protéines de liaison. Répétez cette étape deux fois.
- Effectuer préfractionnement peptide au moyen de chromatographie échangeuse de cations forte à l'aide pas à pas (n = 4) élution à force ionique a augmenté de 19. Séchez le bas des échantillons sous vide en utilisant un concentrateur speedvac.
- Analyser les fractions peptidiques par des moyens de nanodébit chromatographie liquide en phase inverse C18 (1100, Agilent Technologies, Santa Clara, CA) interfacés à électrospray spectrométrie de masse en tandem (LC-MS/MS). Les expériences ont été réalisées sur MS un hybride linéaire iontrap / transformée de Fourier résonance cyclotronique ionique (FTICR) instrument (LTQ FT 7T, Thermo Scientific, Waltham, MA,) Peptide spectres fulscan (m / z 150-2000) Ont été acquises avec l'analyseur FTICR à masse à haute résolution (100 000), suivie par la fragmentation ultérieure des 5 pics peptidiques les plus intenses dans le iontrap par le biais de dissociation induite par collision (CID) 9.
- Identification de la séquence peptidique est réalisée par mise en correspondance base de données et peut être complétée par de novo analyse du séquençage. Pour la recherche de base de données, moteurs de recherche disponibles dans le commerce (Mascot, XTandem ou de la protéine de prospecteur) sont employés 20. Les recherches sont généralement effectuées contre des bases de données contenant des séquences de neuropeptides connus ou prévus et les séquences de protéines précurseurs de neuropeptides 21.
6. Les résultats représentatifs
Spectrométrie de masse MALDI imagerie des coupes de tissus striataux tel que préparé selon le protocole décrit ici conduit à la détection de plus de 1000 pics correspondant à environ 300 mono isotopiques espèces moléculaires moyens indiqués spectresà la Fig. 1). La visualisation des données pour les éminents pics moléculaires a été réalisé avec le logiciel d'imagerie Flex et a montré caractéristiques distributions d'intensité de pointe qui sont bien en ligne avec les caractéristiques anatomiques (Fig. 3). Une autre caractéristique de MALDI IMS est sa reproductibilité relative bonne. Dans cette expérience, le coefficient global de la variance pour les intensités des pics de toutes les espèces détectées moléculaires était de 30%, mais de nombreux pics affiché une variation très faible et une reproductibilité élevée dans les groupes de traitement (Fig. 4). Les données relatives intensité du pic de quatre régions différentes de l'intérêt, y compris la partie dorsolatérale et dorso de striata fois lésé et intact ont été soumis à une analyse statistique. Afin d'ajuster pour les comparaisons multiples simultanément, l'analyse statistique a été réalisée au moyen de tests non paramétriques en utilisant l'outil SAM 18. Les changements les plus importants ont été trouvés dans la partie dorsolatérale de la dopamine-dénervé, le striatum parkinsonien. Ici si changements dans séquent entre les différents groupes de traitement ont été observés pour les peptides dynorphine deux; dynorphine B et alpha-neoendorphin (Fig.5). Dans le détail, une augmentation relative des deux intensités maximales dynorphine de 50-60% a été observée dans les animaux élevés par rapport aux faibles dyskinétiques animaux dyskinétiques et des contrôles des lésions (p <0,05, F (2, 15) = 12,8 DynB, F = 5,7 Aneo; Fig. 5).

. Figure 1 Moyenne MS retrace obtenu à partir de deux régions étroitement liées du striatum; caudé putamen (CPU) et le noyau accumbens (NAc). Les deux régions présentent différents profils de MS avec quelques espèces moléculaires unique exprimées dans une région, ou à différents niveaux d'intensité de pointe (insert, m / z 2028). La répartition spatiale de chaque pic peut être visualisée à l'aide de logiciels d'imagerie spécialisés (panneau inférieur).
2.jpg "/>
Figure 2 (A) Le cerveau est monté sur un mandrin en utilisant un cryostat médias incorporation (OTC; flèche)., On prend soin que l'OTC ne pas contaminer la zone du cerveau à être sectionné depuis la suppression de gré à gré d'ions cause de peptides. (B, C) les articles mince (épaisseur ≈ um 12) sont montés sur le dégel des diapositives MALDI verre compatibles et séché pendant quelques secondes pour éviter d'endommager le gel comme on le voit dans C (D) microfissures peut être difficile à détecter à l'œil nu , mais compromettre matrice MALDI cristallisation et effacer MALDI MS signal. Le même article colorées avec du violet de crésyl révèle microfissures et fissures (en bas à droite microphotographie).

Figure 3. La première étape de l'évaluation des données est de visualiser plusieurs pics différents à travers la gamme de masse analysé (AI). Ici, les articles du striatum de 9 souris ont été imagées avec spectrométrie de masse MALDI. Visualisation du total en moyenne isur le courant va révéler les zones de remarquables intensités ioniques haute ou basse (flèches). Ces zones peuvent être affectées par sur-ou sous-normalisation des effets et de fausser l'analyse des données compromettre les résultats. Pauvre définition anatomique des distributions de pointe révèlent sections, avec un pic généralement faible rapport signal-bruit, par exemple les articles 3 et 9, F pics à I.

Figure 4. Reproductibilité MS entre les groupes de traitement peut être évaluée en calculant la moyenne MS trace et l'erreur-type pour chaque valeur de m / z (inserts, m / z 722 et 1749). Une bonne reproductibilité assure une analyse statistique valable.
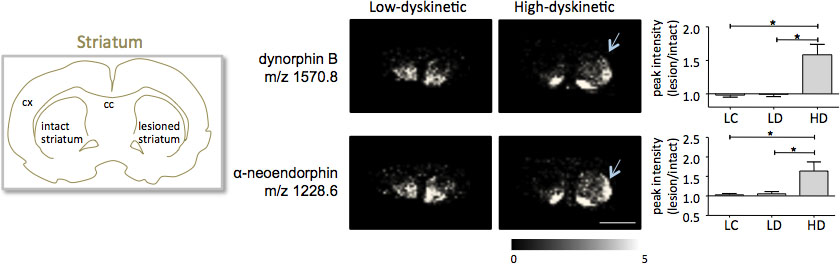
Figure 5. Dynorphine B et alpha-neoendorphin intensités des pics sont significativement augmentés dans le 6-OHDA-Lésé, parkinsonien, le striatum de haute dyskinétiques animaux (HD; flèches) par rapport au groupe témoin à faible dyskinétique (LD) et de la lésion (LC). Intensités des pics peptidiques exprimés en moyenne fois-changement de côté intact ± SEM (lésion / intacte côté). * P <0,05; cx cortex; cc corps calleux. La barre d'échelle 5 mm.
Discussion
Il ya plusieurs avantages à l'emploi de la spectrométrie de masse MALDI imagerie dans l'étude des neuropeptides. Une analyse impartiale des données MS peut révéler que les noyaux du cerveau seulement spécifique, ou, comme dans les résultats présentés ici où seule la partie dorsolatérale du striatum est associée à une certaine condition physiopathologique. En conservant l'information spatiale, il est alors possible de redéfinir les régions présentant un intérêt pour effectuer des analyses statistiques avec une plus grande sensibilité et une plus faible variabilité par rapport à l'analyse de coupes de cerveau entier ou en utilisant les études traditionnelles sur Peptidomics extraits peptidiques. En outre, il est important de réaliser MALDI IMS permet de détecter facilement jusque-là inconnues modifications post-traductionnelles, mais les analyses structurelles doivent suivre pour déterminer les positions exactes des acides aminés qui sont modifiés.
Pièges les plus courants dans la visualisation des données MALDI IMS comprennent la cartographie, l'intensité du pic maximum à une échelle linéaire optique de black (0%) à la couleur (100%) pour chacune des sections de la série expérimentale (Figure 3), au lieu de mapper toutes les sections à une échelle commune absolue, où 100% est le pic d'intensité maximale de toutes les sections (Figure 5) . Cette dernière méthode permet une comparaison des données du groupe et la visualisation des différences entre les groupes de traitement.
Un obstacle majeur à l'analyse MALDI IMS est la cession de peptides à des pics de masse spécifiques. Sur des tissus en tandem de spectrométrie de masse est parfois possible, mais se révèle souvent très difficile 13,14. Nous constatons que l'approche plus traditionnelle comprenant un fractionnement préparatif sur une forte chromatographie par échange cationique, suivi par LC-MS/MS en phase inverse peut être utilisé pour de nombreux neuropeptides séquence avec succès et en particulier des peptides opioïdes. Il est encore pas rare d'obtenir une bonne qualité de spectres MS / MS qui ne correspond pas à toutes les entrées de bases de données en utilisant les moteurs de recherche communs, tels que MASCOT. Dans ces cas de novo-séquençage à la main est l'ONLl'option y. La preuve ultime de l'identité de pointe peuvent être obtenus par MALDI IMS des coupes de tissus de la souris knock-out appropriée, mais ce n'est pas toujours disponible ou possible. Une alternative consiste à valider les résultats par une méthode diamétralement différente, par exemple par immunoblot de l'Ouest ou immunohistochimie. Cela peut souvent inclure des anticorps et une quantité importante de travail de la validation des nouveaux anticorps.
La stratégie générale présentée dans le présent protocole est optimisé pour les grandes expériences à l'échelle du neuropeptide IMS MALDI, y compris plusieurs sections et des conditions expérimentales. Le protocole a été spécialement optimisé pour les peptides opioïdes et auront un impact important dans les études futures, tel qu'il est employé dans divers domaines de recherche, y compris la douleur mécanismes sous-jacents et la réponse endogène à la drogue de dépendance.
Disclosures
Les auteurs n'ont rien à révéler.
Acknowledgments
Nous remercions Hanna Warner pour contribuer les données de la figure 3 et le professeur Jonas Bergquist pour une contribution précieuse. Le Conseil de recherche suédois (Grant 522-2006-6416 (MA), 521-2007-5407 (MA); Fondation Le Wiberg Åke (MA, JH), L'Académie royale suédoise des sciences (MA, JH), et des produits chimiques suédoise Société (JH) sont remerciées pour le soutien financier.
References
- Obeso, J. A., Olanow, C. W., Nutt, J. G. Levodopa motor complications in Parkinson's disease. Trends Neurosci. 23, S2-S7 (2000).
- Caprioli, R. M., Farmer, T. B., Gile, J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. MALDI-TOF MS. Anal. Chem. 69, 4751-4760 (1997).
- Obeso, J. A. The evolution and origin of motor complications in Parkinson's disease. Neurology. 55, S13-S20 (2000).
- O, W. H. Noncommunicable Diseases and Mental Health Cluster, Noncommunicable Disease Prevention and Health Promotion Department, Ageing and Life Course. Active Ageing: A Policy framework. , (2002).
- Schapira, A. H. Movement disorders: advances in cause and treatment. Lancet Neurology. , 6-7 (2010).
- Obeso, J. A., Rodriguez-Oroz, M. C., Rodriguez, M., DeLong, M. R., Olanow, C. W. Pathophysiology of levodopa-induced dyskinesias in Parkinson's disease: problems with the current model. Ann. Neurol. 47, S22-S32 (2000).
- Cenci, M. A., Lee, C. S., Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 10, 2694-2706 (1998).
- Andersson, M., Hilbertson, A., Cenci, M. A. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis. 6, 461-474 (1999).
- Hanrieder, J. Alterations of striatal neuropeptides revealed by imaging mass spectrometry. Molecular & Cellular Proteomics. , (2011).
- Cornett, D. S., Reyzer, M. L., Chaurand, P., Caprioli, R. M. MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods. 4, 828-833 (2007).
- Ljungdahl, Imaging Mass Spectrometry Reveals Elevated Nigral Levels of Dynorphin Neuropeptides in L-DOPA-Induced Dyskinesia in Rat Model of Parkinson's Disease. PLoS ONE. 6, e25653 (2011).
- Groseclose, M. R., Andersson, M., Hardesty, W. M., Caprioli, R. M. Identification of proteins directly from tissue: in situ tryptic digestions coupled with imaging mass spectrometry. J. Mass. Spectrom. 42, 254-262 (2007).
- Andersson, M., Groseclose, M. R., Deutch, A. Y., Caprioli, R. M. Imaging mass spectrometry of proteins and peptides: 3D volume reconstruction. Nat. Methods. 5, 101-108 (2008).
- Deininger, S. -O. Imaging Mass Spectrometry. Setou, M. , Springer. Japan. 199-208 (2010).
- Norris, J. L. Processing MALDI Mass Spectra to Improve Mass Spectral Direct Tissue Analysis. Int. J. Mass. Spectrom. 260, 212-221 (2007).
- Ihaka, R., Gentleman, R. R. A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics. 5, 299-314 (1996).
- Mass Spectrometry Binning Software GAB. , Vanderbilt Center for Quantitative Sciences. Nashville, TN. Available from: http://www.vicc.org/biostatistics/software.php (2012).
- Tusher, V. G., Tibshirani, R., Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116-5121 (2001).
- Bergstrom, L., Christensson, I., Folkesson, R., Stenstrom, B., Terenius, L. An ion exchange chromatography and radioimmunoassay procedure for measuring opioid peptides and substance P. Life. Sci. 33, 1613-1619 (1983).
- Falth, M. Neuropeptidomics strategies for specific and sensitive identification of endogenous peptides. Mol. Cell. Proteomics. 6, 1188-1197 (2007).
- Falth, M. SwePep, a database designed for endogenous peptides and mass spectrometry. Mol. Cell. Proteomics. 5, 998-1005 (2006).
