1. Culturing of the Macrophage Cell Line RAW 264.7
- Maintain RAW 264.7 cells (ATCC, catalog # TIB-71) in DMEM-10 media, which consists of high glucose DMEM with 10% (v/v) low-endotoxin fetal bovine serum (< 10 EU/ml), 10 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM non-essential amino acids, 2 mM L-glutamine. Cells are typically maintained in 175 cm2 tissue culture flasks containing 35 ml of media per flask and incubated at 37 °C and 5% CO2 in a tissue culture incubator. However, any size flask can be used with a volume of media that is appropriate for the size of the flask.
- To split cells: aspirate off the old media, add 10 ml of fresh DMEM-10 media to the cells, and then scrape the cells from the bottom of the flask by using a cell scraper. Next, resuspend the cells into a homogenous solution by drawing up cells into a 10 ml pipette and forcefully squeezing the cells through the pipette tip pressed against the bottom of the flask. Repeat this action at least 3 times so the cells no longer clump together. Verify by light microscopy that a single cell suspension was generated. Then transfer 1 ml (1:10 dilution or ~1×107 cells) – 2 ml (1:5 dilution or ~2×107 cells) of the cell suspension to a new 175 cm2 flask, and bring the final volume of media up to 35 ml.
- Split cells when they are nearly confluent (~1×108 cells total/175 cm2 flasks): every three days if starting with a 1:10 dilution, or every two days if starting with a 1:5 dilution. Use light microscopy to check cell morphology before splitting cells. Most of the cells should look round and not activated. Activated cells have granules and/or extended, spindly morphology with appendages. Do not let cells overgrow as those cells do not typically form plaques. Keep track of the passage number and frequently start over by thawing a lower passage aliquot of cells. (We use passage 30 as a cut-off).
2. Infect RAW 264.7 Cells with MNV Inoculum
- Seed RAW 264.7 cells into 6-well plates (3.5 cm diameter) at a density of 1×106 viable cells/ml in DMEM-10 media, and add 2 ml of this suspension to each well. It is important to distribute cells evenly in wells either by rocking plates by hand at least 10 times or by using a rocking apparatus for ~10 min. Do not swirl the plates as this will cause the cells to cluster in the center of the well. Place plates into a tissue culture incubator (at 37 °C and 5% CO2). Allow cells to attach overnight or for at least 4 hr at 37 °C. Cells should be 60 – 80% confluent for the plaque assay and distributed evenly throughout the well.
- The next day, prepare the virus inoculum, which can be from MNV-infected cells in tissue culture or from homogenized tissues or fecal samples of MNV-infected mice. When using tissue samples, pea-sized pieces of tissue are homogenized in 2 ml screw-cap tubes containing sterile silica beads in 1 ml of DMEM-10 using a tissue homogenizer (e.g. MagnaLyser; Roche). For fecal samples, no more than 3 fecal pellets should be homogenized in 1 ml media. All samples are then frozen (at -80 °C) and thawed once before performing the plaque assay.
- Prepare 10-fold dilutions of the virus inoculum in complete DMEM-5 medium, which consists of DMEM/High glucose, 5% (v/v) low-endotoxin fetal bovine serum (< 10 EU/ml), 10 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM non-essential amino acids, 2 mM L-glutamine.
- Ten-fold serial dilutions are prepared in 24-well plates: A repeater pipette is used to dispense 1.35 ml media into multiple wells, the 10-1 dilution is made by mixing 1.35 ml of media and 0.15 ml of virus-containing sample, and then 0.15 ml of the 10-1 dilution is added to 1.35 ml of media to make the 10-2 dilution and so on. It is important to change tips each time you make a new dilution. A multichannel pipette can be used to make the dilutions of multiple samples at a time with two tips fitting into one well of a 24-well plate transferring a total volume of 0.15 ml per well (see Figure 3A).
- A typical dilution range for tissue homogenates and fecal contents is 10-1 to 10-3. However, plaques from these samples tend to be smaller compared to those from tissue culture samples. Furthermore, in some cases a 1:100 dilution of fecal samples is needed to sufficiently dilute out any toxic components of the feces that may disrupt the cell monolayer, thus hindering the ability to count plaques. The dilution range of tissue culture lysates depends on the time point of interest during the viral life cycle. Dilutions that go up to 10-9 may be needed at the peak of infection.
- After the serial dilutions are prepared, label the 6-well plates containing RAW 264.7 monolayers (from section 2.1) with the sample name and dilutions being plated. One plate at a time, remove all media by flicking it out or aspirating it. Immediately afterwards add 0.5 ml of a diluted sample to a well, then repeat with a duplicate well, before proceeding to the next dilution. Once all 3 dilutions are added to one plate, tilt plate back and forth by hand to ensure all cells have been covered. Handle one plate at a time to ensure that cells will not dry out.
- After adding 0.5 ml of the dilutions to each well, stack plates upright and incubate them for 1 hr at room temperature. Because the volume added to each well is not sufficient to cover the monolayer completely, the plates need to be gently tilted back and forth by hand every 10-15 min or placed on a rocking apparatus (~18 oscillations per min). This prevents cells from drying out.
3. Low Melting Point Agarose (SeaPlaque) Overlay Preparation
Note: it is advisable to have several bottles with autoclaved SeaPlaque agarose prepared ahead of time. Agarose can be re-melted in a microwave before use.
- Calculate the amount of overlay required for the total volume of plates before the 1 hr incubation is complete. The volume needed is 2 ml/well or 12 ml/6-well plate. Prepare agarose (see section 3.2) and media (see section 3.3) separately.
- To prepare the agarose, suspend 3 g of SeaPlaque agarose in a total volume of 100 ml of distilled water (3% w/v) in a glass bottle. Autoclave for 20-30 min. (If agarose was already prepared before-hand, re-melt agarose in microwave.) It is important to equilibrate SeaPlaque agarose to 42 °C in a water bath before use because if the agarose is too hot, it will kill the cells. Make sure water level is equal to or above the level of the agarose to avoid undesired solidification.
- To prepare the media: make 100 ml of 2x MEM media, which consists of 2x MEM, 10% (v/v) low-endotoxin fetal bovine serum (< 10 EU/ml), 10 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, 4 mM L-glutamine. Equilibrate media to 37 °C in a water bath.
- Mix both the SeaPlaque agarose and the 2x MEM media together in a sterile bottle at a 1:1 ratio immediately before overlaying the infected cell monolayers. If more than 200 ml overlay is needed, split volume into multiple bottles and keep in 37 °C water bath until ready to use.
- At the end of the 1 hr incubation (see section 2.7), aspirate the inoculum off of each well. Slowly add 2 ml of overlay to the edge of each well by placing the pipette tip against the wall of each well. Up to 5 plates can be handled simultaneously without cells drying out.
- Allow the overlay to solidify for approximately 10 min at room temperature before placing plates upright into the tissue culture incubator. Incubate plates for 48 hr at 37 °C in 5% CO2.
- After the incubation period, plaques are faintly visible to the naked eye, so check unstained plates for presence of plaques. If no plaques are visible, incubate for an additional 4 hr and check again. However, the maximum incubation time should not exceed 72 hr.
4. Visualization of Plaques by Neutral Red Staining
- To visualize plaques, the neutral red staining solution is prepared by adding 3 ml of neutral red (0.33% w/v in DPBS; Sigma, catalog # N2889) to every 97 ml of 1x PBS (tissue culture grade, Mg2+-, Ca2+-free; Gibco, catalog # 10010). Calculate the volume of neutral red staining solution needed for the experiment: 12 ml neutral red staining solution are required for each 6-well plate. Then, add 2 ml to each well. Although some plaque assay protocols require the agarose plug to be removed from the wells, in this protocol the neutral red staining solution is added directly onto the overlay.
- After a one hr incubation at 37 °C, check if plaques are visible with neutral red staining solution still in wells. If plaques are not readily apparent, allow the staining to continue for another hr. Continue incubating until plaques are visible. (Note: Staining for more than 3 hr is not optimal and if no plaques are visible in the positive control sample after 3 hr of staining, the plaque assay did not work properly.) After the staining is complete, aspirate the neutral red staining solution, ensuring the agarose plug is not disturbed, and then proceed to counting the plaques.
- Count plaques by placing plate upside down on a light box and marking a dot on counted plaques to avoid duplicate counts. Choose the dilution to count plaques in wells where plaques are clearly separated (i.e. no visual evidence of plaques fusing together). If possible, count plaques at two dilutions. It is important to note that plaque size may vary between MNV strains, virus inoculum, and depends on the condition of the RAW 264.7 cells during the plaque assay.
- If no plaques are visible in a well, either there was no virus present in the sample or the amount of virus was under the limit of detection of the plaque assay. In this case, the wells stain red with a similar color as other plaque-containing wells. Alternatively, the absence of plaques is also observed when there are too many viral particles present in a given dilution. This leads to lysis of the entire monolayer and wells appear orange/yellow in color.
- Calculate viral titers. Add the number of plaques in both wells at a single dilution and multiply by the dilution factor (i.e. 1 ml if 2 wells are infected with 0.5 ml). This will yield the amount of plaque forming units (PFU) in your inoculum volume of 1 ml. For example, in Figure 4 at the 10-2 dilution, one well (marked “II”) has 14 plaques and the other well (marked “V”) has 17 plaques. Thus, the viral titer will be 14×102 + 17×102 = 3,100 (3.1×103) pfu/ml.
5. Representative Results
Infectious MNV-1 particles can be quantified using a plaque assay as outlined schematically in Figure 1. Figure 2A shows a well with a monolayer of RAW 264.7 cells just prior to infection, while Figure 2B shows three visible plaques indicated by roman numbers I, II and III in a well. Individual steps of the assay are depicted in Figures 3A through F. Figure 3A shows the preparation of the 10-fold dilution series of a virus-containing sample. Figure 3B shows the transfer of dilutions to duplicate wells of a 6-well plate. Figure 3C shows the rocking apparatus used to incubate RAW 264.7 cells with the inoculum at room temperature for 1 hr. Figure 3D shows cells being overlaid with the SeaPlaque:MEM mixture. Figure 3E shows a plate at room temperature to allow the overlay to solidify, while Figure 3F shows cells being stained with a 0.01% neutral red solution 48 hr later. After staining cells for 1-3 hr and aspirating the neutral red staining solution, plaques are visible and can be counted (Figure 4).

Figure 1. Schematic of the MNV plaque assay protocol.
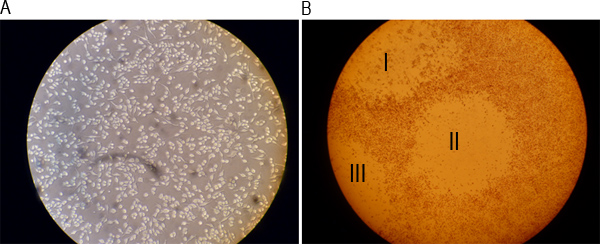
Figure 2. Representative images of a well of a monolayer before infection and after formation of plaques. A) RAW 264.7 cells were cultured overnight and imaged under a light microscope at 20x magnification. B) Cells were stained with a 0.01% neutral red solution after 48 hr of infection and visualized under a light microscope at 4x magnification. Roman numbers I, II, and III indicate three visible plaques.

Figure 3. Representative images of the different plaque assay steps. A) MNV-1 inoculum is prepared in 10-fold dilutions. B) Inoculum is added to cell monolayers in duplicate wells. C) Cells and inoculum are incubated by rocking for 1 hr at room temperature. D) Cells are overlaid with a 1:1 mixture of SeaPlaque agarose and 2x MEM media. E) Plates are incubated for 10 min at room temperature to allow the overlay to solidify. F) Staining of cells with the neutral red staining solution 48 hr post-infection.

Figure 4. MNV-1 forms plaques in cell monolayers. Shown here is a representative plaque assay plate 48 hr post-infection, showing plaques stained with neutral red staining solution after 1 hr of incubation. The plate shows duplicate wells of three 10-fold dilutions. Wells labeled with roman numbers I and IV correspond to the 10-1 dilution; II and V correspond to the 10-2 dilution; III and VI correspond to the 10-3 dilution. The viral titer of the sample is indicated below (see Section 4.5 for details of the calculation).
