

Summary
Vi beskriver de bildedannende metoder vi bruker for å undersøke fordelingen og bevegelsesfrihet av de transfekterte fluorescerende proteiner resident i det endoplasmatiske retikulum (ER) ved hjelp av konfokal avbildning av levende celler. Vi har også ultrastructurally analysere effekten av sitt uttrykk på arkitekturen i denne subcellulære rommet.
Abstract
De lipider og proteiner i eukaryotiske celler er kontinuerlig utvekslet mellom cellekamre, selv om disse beholder sin karakteristiske sammensetning og funksjon til tross for den intense interorganelle molekyl trafikk. Teknikkene som beskrives i denne artikkelen er kraftige virkemidler for å studere protein og lipid mobilitet og trafficking in vivo og i deres fysiologiske miljø. Fluorescens restitusjon etter photobleaching (FRAP) og fluorescens tap i photobleaching (FLIP) er mye brukt direkte-celle avbildningsteknikker for å studere intracellulær trafikkering gjennom exo-endocytoseveien, kontinuiteten mellom organeller eller subcompartments, dannelsen av proteinkomplekser og protein lokalisering i lipid microdomains, kan alle bli observert under fysiologiske og patologiske tilstander. Begrensningene av disse metodene er i hovedsak på grunn av bruk av fluoriserende fusjon proteiner, og deres potensielle ulemper inkluderer kunstig over-expression i celler og muligheten for forskjeller i folding og lokalisering av kodede og native proteiner. Til slutt, som grense for oppløsning av optisk mikroskopi (ca. 200 nm) ikke tillater undersøkelse av den fine struktur av ER eller de spesifikke subcompartments som kan stamme fra cellene under stress (dvs. hypoksi, legemiddeladministrering, på over-ekspresjon av transmembrane ER bosatt proteiner) eller under patologiske tilstander, kombinerer vi live-cell avbildning av dyrkede transfekterte celler med ultrastructural analyser basert på transmisjonselektronmikroskopi.
Introduction
Oppdagelsen av grønt fluorescerende protein (GFP) og dets spektrale varianter, og den parallelle utviklingen av fluorescens mikroskopi, har åpnet opp helt nye muligheter for etterforskningen av protein atferd i cellene. Teknikker slik som fluorescens gjenvinning etter photobleaching (FRAP) og fluorescens tap i photobleaching (FLIP), noe som er mulig på grunn av den iboende kapasitet av fluoroforer til å slukke deres fluorescens under intens belysning, er basert på konfokal levende celler avbildning og bruk av transfekterte fluorescerende fusjon proteiner 1-3. De er mye brukt for å vurdere ikke bare lokalisering av proteiner, men også deres mobilitet og vesikulært transport, som kan avsløre viktige ledetråder om deres funksjon 4.
Det unike med eukaryote celler er tilstedeværelsen av intracellulære rom som har bestemte lipid-og proteinsammensetningen. Selv om organeller er fysisk isolated, at de trenger å kommunisere med hverandre og dele molekylære komponenter for å opprettholde cellulær homeostase. Eliminasjonen garanterer at proteiner og lipider syntetisert i ER når den riktige endelige bestemmelsessted hvor de utøver sin funksjon. Intracellulære organeller kan også være forbundet med dynamiske kontaktsider som tillater molekyler (lipider) som skal direkte vekslet mellom avdelinger. Videre har mange proteiner har til sammen i store heteromeric komplekser eller knyttet til bestemte lipid arter (lipid flåter / microdomains) for å bli funksjonelt aktiv eller å bli fraktet til sin endelige destinasjon. Alle disse biologiske aspekter i stor grad påvirker de kinetiske egenskaper av proteiner, og kan derfor være hensiktsmessig undersøkes ved hjelp av de teknikker som er beskrevet nedenfor.
Vår gruppe har mye brukt FRAP og Flip kombinert med elektronmikroskopi for å studere arkitektur av ER og dens forskjellige subdomener. ER er den første stasjonen i sekresjonssporet og spiller en nøkkelrolle i protein og lipid sortering fem. Det er en svært dynamisk organeller som tydelig underdomener reflektere sine mange forskjellige funksjoner (dvs. protein og lipid biosyntese og menneskehandel, protein folding, Ca 2 + lagring og slipp, og xenobiotic metabolisme). Imidlertid, selv om de er morfologisk, romlig og funksjonelt distinkte, disse domenene er kontinuerlige med hverandre, og deres relative overflod kan modifiseres på celler under fysiologiske og patologiske tilstander. Den best kjente og vanligvis romlig atskilte områder av ER er atom konvolutten, og glatt og ru ER, men har vi og andre har vist at det er ER strukturer med en mer forseggjort arkitektur og tredimensjonale organisasjon i forskjellige celletyper, og vev under fysiologiske betingelser, som også kan induseres ved hjelp av stressende stimuli som hypoksi, medikamentadministrasjon, eller over-uttrykk for ER-resident transmembrane proteiner 2,6 (og referanser deri).
Vi har også nylig vist nærværet av slike strukturer i cellemodeller av humane sykdommer 1,7. Stammer fra den stablet cisternae av glatt ER, ble de gitt samlebetegnelsen for organisert glatte endoplasmatiske retikulum (OSER) i 2003 6, selv om de er også kjent som karmellae, lameller, og Krystalloide ER på grunnlag av sin arkitektur som i likhet med sin størrelse, kan variere. Etter at cellene er transfektert med GFP kondensert til den cytosoliske region av tail-ankret (TA) ER-resident proteiner (d EGFP-ER), den svakt dimerizing tendensen av GFP i trans endrer dramatisk organiseringen og strukturen av ER. FRAP og FLIP eksperimenter viste at d EGFP-ER er gratis å spre innenfor OSERs, og det faktum at den beveger seg fra den retikulære ER til OSER og vice versa </ Em> viser at aggregatene er kontinuerlig med den omkringliggende retikulære ER. Ultrastructural analyse har tillatt oss å relatere fluorescens data med en detaljert beskrivelse av OSER arkitektur og organisasjon på nanoskala nivå: OSERs er alltid laget opp av stabler av paret cisternae av glatt ER, men kan ha ulike former for romlig organisering, for eksempel jevnlig arrangert sinusformet matriser eller spinnehjul, eller sekskantede "krystalloide" tubular arrays. Disse rearrangements føre til kubikk morfologi 8 som, som de har blitt funnet i celler under fysiologiske forhold 9 og følgende påkjenninger som hypoksi 10, medikamentell behandling 11, og kreft 9, kan ha betydelig potensial som ultrastructural markører.
Etter denne første demonstrasjonen ved hjelp av GFP fusjon proteiner, brukte vi bildebehandling eksperimenter å analysere spredning av ER domener som svar på farmakologisk behandling 12, assess tendensen av fluorescerende proteiner til oligomerise i cellene 13, og for å undersøke hvilken rolle en mutant, ALS-linked TA protein i dannelsen av intracellulære aggregater av ER opprinnelse som kan være relevante for sin patogenitet 1,8. Det har blitt foreslått at dannelsen av intracellulære aggregater (som forekommer i mange nevrodegenerative sykdommer 14) kan være en beskyttende mekanisme utformet for å forhindre interaksjoner mellom toksiske mutante proteiner og de omliggende cellekomponenter 15.
Det som følger er en beskrivelse av en kombinasjon av optiske og elektronmikroskopi metoder for å undersøke konstruksjoner som C-terminal hydrofobe domener er satt inn i membranen av akuttmottaket, og en analyse av deres dynamiske oppførsel og virkningene av deres overuttrykk på ER domene Arkitekturen i dyrkede celler (se figur 1 for et flytskjema av den eksperimentelle protokollen).
Subscription Required. Please recommend JoVE to your librarian.
Protocol
En. Plasmid, Cell Culture, og Transfection med ER Fluorescent Proteiner
- Plasmidet som anvendes i denne studien består av en forbedret versjon av GFP smeltet ved sin C-terminus til halen region av ER isoformen av rotte cytokrom b (5) (forkortet her som b (5)) via en linker-sekvens. Halen regionen inneholder hele sekvensen (Pro94-Asp134) som forblir membranassosiert etter trypsin-spalting av nativt b (5), blant annet av 17-residuet TMD (transmembrane domene), flankert av oppstrøms-og nedstrøms polare sekvenser (UPS og DPS) . Linkeren består av myc epitopen fulgt av [(Gly) 4 Ser] 3, og det hele cDNA blir satt inn i de Hind3-Xba1 områder av pattedyr ekspresjonsvektor pCDNA3. Detaljene i konstruksjonen av dette plasmid er beskrevet i en tidligere publikasjon hvor det er referert til som GFP-ER 16.
- Dyrke COS-7-celler i Dulbeccos modifiserte Eagle Medium (DMEM) supplert med 10% fetal bovint serum, 2 mM L-glutamin, 1% penicillin / streptomycin i en inkubator ved 37 ° C og 10% CO2.
- Transfeksjon. Plate 3 x 10 5 celler på en rund glass dekkglass i en 6-brønns plate og, på den følgende dag, transfektere med JetPEI system som beskrevet av produsenten. Legg merke til at det optimale JetPEI / DNA-forhold har blitt testet for å etablere maksimal transfeksjon effektivitet avhengig av plasmid-og cellelinje som brukes: i vårt tilfelle, en JetPEI: DNA ratio på 2:1 fører til 70-80% transfeksjon effektivitet.
2. Live-fluorescens Scanning konfokalmikroskopi
- Live-cell imaging. Sett på dekkglass på hvilken de transfekterte celler ble utsådd i en stål kulturcellekammer for 24 mm dekkglass som er fylt med DMEM w / o fenol rød, supplert med 10% FBS, 2 mM L-glutamin, 1% pen / strep, 25 mM HEPES , 50 pg / ml cykloheksimid og 1:100 OxyFluor å hindre at prøvene fra fotobleking. En SP5 confocal mikroskope er utstyrt med en temperaturkontrollert CO 2 inkubator (37 ° C og 5% CO 2) brukes for levende celle avbildnings eksperimenter, med d GFP-ER blir visualisert ved å bruke en 488 nm laser og en 525/50 båndpassemisjonsfilter.
- Fluorescens restitusjon etter photobleaching (FRAP). Tegn en region av interesse (ROI), tilsvarende en OSER struktur, og bleike det bruker 20 gjentakelser og en kombinasjon av 488 nm (100% av en 30 mW Argon laser, tilsvarende 5,5-6 μW på prøven) og 405 nm (60% av en 30 mW Diode 405 laser, tilsvarende 11,6 μW på prøven) lasere som etter vår erfaring, fører til effektiv og rask fotobleking.
- Spill utvinning av fluorescens i de bleket Rois ved å ta ett enkelt bilde hver 10 sek for 10 min (pixel tid = 1,61 usekunder / px).
- Fluorescens tap i fotobleking (FLIP). Tegn en ROI tilsvarende en OSER struktur, og blekemiddel som beskrevet ovenfor. Bleke gjentas hver 30 sek,og post-blekebilder opp hvert 10 sek for 30 min (pixel tid = 1,61 usekunder / px).
- FRAP og FLIP analyse. Alle bildene er analysert ved hjelp ImageJ programvare ( http://rsbweb.nih.gov/ij/download.html ). I FRAP eksperimenter, blir fluorescens utvinning av bleket ROI målt over tid og normalisert til den totale fluorescens av bleket celle, som alltid er kontrollert til å være konstant over tid.
- For de FLIP eksperimenter, tegne en ROI utenfor bleket OSER og dekker hele cellen. Måle fluorescens intensitet over tid, og for å normalisere de fluorescens nivåer av en ROI trukket på et ubleket celle for å korrigere for en hvilken som helst reduksjon i fluorescens forårsaket av avbildning i seg selv.
- I alle forsøkene, trekkes bakgrunnssignalet (bestemt i et område utenfor cellene) fra de fluoriserende intensiteter av ROIs. Til slutt, plott inn resultatene ved hjelp av GraphPad Prism programvare.
Tre. Ultrastructural Analyse ved hjelp av transmisjonselektronmikroskopi
Gitt giftigheten av mange av reagensene, bør alle de fremgangsmåter utføres på seg et passende laboratorium. frakk og hansker under en avtrekkshette.
- Etter fjerning av dekkglass fra petriskålen, fastsette de gjenværende celler dyrket på bunnen av fatet som et monosjikt ved hjelp av filtrert 2% glutaraldehyd i 0,1 M kakodylat-buffer, pH 7,4, i 10 min ved romtemperatur.
- Skrap cellene ved hjelp av en Teflon skrape og overføre dem til 1,5 ml Eppendorf-rør. Pellet cellene ved hjelp av sentrifugering ved 9000 g i 10 min. Fjern supernatanten, tilsett friskt fikseringsmiddel, og la over natten ved 4 ° C.
- Vask pelleten med bufferen, og deretter etter fastsette med en oppløsning av 1% osmium tetroksyd i kakodylat-buffer i 1 time ved romtemperatur.
- Skyll med MilliQ vann, og en bloc flekken med1% uranyl-acetat i destillert vann i mellom 20 til 60 min.
- Dehydrer prøvene i økende etanolserier (70%, 80%, 90%, 100% og 100% i 10 minutter hver), og vaskes kort to ganger etter propylenoksyd (15 min hver).
- Infiltrere prøvene i en blanding av propylenoksyd + Epon (1:1) (fra 2 timer til over natten).
- Bygge inn i Epon epoksyharpiks herdet ved 60 ° C i minst 24 timer.
- Seksjon de manuelt trimmet harpiks blokkene ved hjelp av en ultramicrotome LEICA UC6 utstyrt med en 45 ° diamant kniv til å skaffe deler med en tykkelse på 60-70 nm. Samle avsnittene om 300 mesh kobbernett.
- Flekk seksjonene på risten med en mettet løsning av uranyl-acetat (20 min) og bly-citrat (7 minutter), vaskes grundig ristene ved å dyppe dem i bi-destillert vann, og tillate dem å tørke ved romtemperatur.
- De fargede nett er observert ved hjelp av en Tecnai G2 overføring elektronmikroskop, og bildene er tatt med en bottom-montert CCD-kamera på ulike slutt forstørrelser (generelt spenner fra 6,000-39,000 X).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Figur 2 viser et eksempel FRAP studie av protein mobilitet. Mobiliteten av d EGFP-ER protein er demonstrert av den raske fluorescens restitusjon etter photobleaching i bleket OSERs. For den kvantitative analysen ble den halve tid og mobile fraksjon avledet fra eksperimentelt målte data ved å montere følgende monoexponential ligning:
F (t) = F innlegget + (F rec-F innlegg) (1-e-t / τ)
der F innlegget er fluorescens signal etter fotobleking, er F rec maksimal fluorescens utvinning verdi som kommer etter bleking, t tidspunktet for registrering og τ tidskonstanten.
Vær oppmerksom på viktigheten av å skaffe bilder uten mettet piksler som kan endre fluorescens utvinning og følgelig protein mobilitet analyse. Det jegs også viktig for alltid å normalisere den fluorescens-signal i den blekede ROI til den totale fluorescens i den samme cellen, for å vurdere fluorescensintensitet variasjoner på grunn av bleking under bildeopptak eller små endringer i fokuseringsplanet.
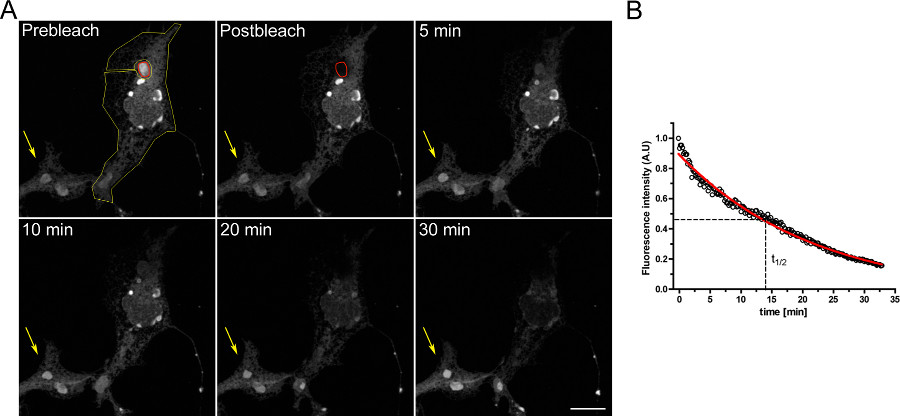
Et eksempel på et FLIP eksperiment for å studere kontinuiteten mellom den intracellulære rom er vist i figur 3.. OSERs er fysisk forbundet med resten av ER som demonstrert ved gradvis tømming av ER når OSER domenet kontinuerlig bleket.
For en skikkelig analyse, må kjøpet av mettet piksler unngås (se ovenfor), og videre, må oppkjøps parametere settes opp med laser krefter så lavt som mulig for å unngå fotobleking grunn image oppkjøpet. Av denne grunn er det sterkt anbefalt å avbilde et ubleket celle i det samme felt som skal brukes til å normalisere den fluorescens-signal til den blekede cell.
Alle eksperimenter må utføres i nærvær av cykloheksimid, et oversettelses inhibitor, for å unngå noen økning i ER fluorescens-signalet (og følgelig total fluorescens) på grunn av protein biosyntese.
Transmisjonselektronmikroskopi viste at fluorescerende aggregater observert i dyrkede celler transfektert med d EGFP-ER representerer flekker av glatt og flatet ER cisternae som romlig organisert seg inn i veldefinerte 3D geometrier klassifisert på grunnlag av sine mønstre: lineære eller buede Søyler (ofte assosiert med kjernefysiske konvolutten, ikke vist) (Tall 4A og B) som kan være kontinuerlig med regioner i sinusformet ER (Figur 4A), membraner i enkelte regioner er organisert i innhegninger med en firkantet eller sekskantet symmetri (Krystalloide ER, ikke vist ). Tilliggende cisternae er adskilt av et tynt lag av svakt elektrontett cytoplasma ca 11 nm tykk som er kontinuerlig med cytoplasma rundt aggregatene.

Figur 1. Flytskjema for den eksperimentelle prosedyre. De dyrkede cellene blir først transfektert med jetPEI (se Protocol) for å over-uttrykke det fluorescerende fusion protein av interesse. Etter 24 timer, er de levende transfekterte celler visualisert og FRAP og FLIP eksperimenter utføres ved hjelp av en konfokalmikroskop utstyrt med en kontrollert temperatur CO 2 inkubator, og de innspilte bilder eksporteres og analysert ved hjelp av egnet programvare (f.eks ImageJ). For ultrastructural analyse, er de transfekterte cellene fast, pelletert og forankret i EPON epoksyresinprodukter blokker. Ultratynne seksjoner er oppnådd ved bruk av en diamantkniv, samlet på copper nett, og observert under en overføring elektronmikroskop. Klikk her for å se større figur .

Figur 2. FRAP eksperiment ved hjelp av COS-7 celler forbigående transfektert med d EGFP-ER. A) To Oser strukturer (røde Rois) ble bleket, og fluorescens utvinning ble registrert over tid. Clear fluorescens utvinning kan bli detektert 1 min etterbleking, og signalet ytterligere økninger 4 minutter senere (skala bar 10 mikrometer) B):. Kvantitativ analyse av FRAP eksperiment som viser utvinning halv-time og den mobile brøkdel av d EGFP -ER protein. Click her for å se større figur

Figur 3. FLIP eksperiment ved hjelp COS-7 celler forbigående transfektert med d EGFP-ER. A) Den kontinuerlige bleking av en OSER (angitt med den røde ROI) fører til en progressiv reduksjon i fluorescens i resten av ER og i andre OSER strukturer innenfor den samme cellen (den gule ROI). Den gule pilen angir en del av en ubleket celle hvor fluorescens-signalet er konstant over tid. (Scale bar 10 mikrometer). B) Kvantitativ analyse av FLIP eksperiment. Klikk her for å se større figur
Figur 4. Etter fiksering og forankring, ble celler som uttrykker høye nivåer av d EGFP-ER hvori OSER strukturer vil kunne detekteres ved hjelp av fluorescens-mikroskopi optiske observeres gjennom et transmisjonselektronmikroskop. A) Lav forstørrelse av en del av cytoplasma til en celle inneholdende en OSER bestående av stablet cisternae og bølgeformede sinusformede membraner. Mitokondrier (M) kan sees gruppert rundt Oser strukturer, mens ribosomer dekorere bare membraner i den ytterste cisternae (pilspisser og innfelt). Den 11 nm tykk elektron-tett mellomrom mellom membraner er kontinuerlig med cytoplasma (pil og innfelt) (L = lysosomes / (auto-) phagosomes) (skala bar 1,5 mikrometer, innfelt 0,25 mikrometer). B) En OSER kan dannes etter blad ER: dvs. stabler av flat ER cisternae som kan være kontinuerlig eller fragmented i sitt utseende i tynne snitt. Blemmer spirende fra den ytterste cisternae av stabelen observeres sporadisk (stjerne) (PM, plasma membran) (Scale bar 150 nm).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Protokollene og bildebehandling tilnærminger beskrevet i denne artikkelen har blitt brukt til å undersøke utbredelse og mobilitet av transfekterte TA fluorescerende proteiner bosatt i ER av levende celler. Vi har også analysert virkningen av over-ekspresjon av disse proteinene på arkitekturen av denne subcellulære rommet ved hjelp av ultra analyser.
Kombinasjonen av live-cell confocal bildebehandling og elektronmikroskopi representerer er et veldig kraftig middel til å undersøke de dynamiske egenskapene til proteiner, og kan gi viktig informasjon om protein funksjon. De beskrevne metoder er ikke tidkrevende (typisk tre dager med arbeid), og utvikling av mange brukervennlige programmer for bildeopptak og analyse gjør photobleaching-basert, live-cell imaging relativt enkel.
Den viktigste begrensning av disse teknikkene er bruken av fluorescerende fusjonsproteiner fordifluoriserende kode kan påvirke riktig folding og / eller montering av proteinet av interesse. Dessuten kan overekspresjon endre oppførselen til transfekterte, fluorescensmerket kodede proteiner, og kan derfor ikke gjenspeile de virkelige egenskaper ved endogene proteiner, men kan dette overvinnes ved anvendelse av induserbare og stabilt transfekterte celler i hvilke uttrykket nivået kan være nøyaktig modulert for å oppnå nivåer sammenlignbare med de av det endogene protein 1,7. Tendensen med fps til oligomerise har vært mye dokumentert og kan vesentlig endre atferd (dvs. kinetikk, uønskede protein-protein interaksjoner og dannelse av aggregater) av kimære proteiner. Bruken av optimaliserte monomere fluorescerende proteiner bør derfor vurderes 17.
Et annet viktig aspekt av dynamiske bildediagnostikk ved hjelp av fluorescens og fotobleking er den tiden som trengs for å bleke fluorescens effektivt og måle fluorescens recovery (og således proteinet mobilitet) presist, noe som også er avhengig av det området av ROI og lokal celletykkelse. Dersom en gitt GFP-merket protein har en høy diffusjonskoeffisienten, kan diffusjon forekommer i løpet av blekingen og dermed forstyrre utvinning tidsmålinger. For å oppnå hurtig og effektiv bleking, er det sterkt anbefalt at en "zoome inn"-funksjon (hvis tilgjengelig) og mer enn én laserlinjen benyttes. Selv om bruken av en hurtig skanning modul (dvs. en resonans-scanner) kan forbedre hastigheten for avbildning under utvinning fase av et eksperiment, i våre hender det også i betydelig grad reduserer blekeeffektivitet. Imidlertid kan alternative skanning systemer (for eksempel en roterende plate utstyrt med en dedikert fotobleking enhet), og mer kraftige lasere forbedre både blekeeffektiviteten og oppkjøp hastighet.
De fleste fluorescerende proteiner som brukes i FRAP og FLIP eksperimenter viser en viss grad av reversibel fotobleking og BLInking som må vurderes når du utfører kvantitative analyser. Svingningene mellom fluorescerende og mørke tilstander forekommer i andre til minutt tidsskala. For EGFP, har det vist seg at under blekeeksperimenter, kan fluorescens variasjoner medfører mindre enn 10% av molekylene, altså i det foreliggende protokoll dette fenomenet er ubetydelig. Hvis alle betingelser holdes konstante, vil dette innføre en konstant skjevhet i resultatene. Hvis andre fluorescerende proteiner blir brukt, der reversible brøkdel er betydelig høyere (dvs. YFP), eller å oppdage og vurdere photobleaching reversibilitet, kan dette gjøres ved å måle fluorescens gjenoppretting etter fotobleking i hele levende celle, dersom gjenvinning er observert dette kan bare være et resultat av fotobleking reversibilitet 18.
Den potensielle toksisitet av lys under forsøkene er en annen viktig faktor, særlig fordi fotobleking krever sterk belysning. Det er well kjent at eksiterte fluoroforer en tendens til å reagere med oksygen for å produsere frie radikaler, som kan påvirke forskjellige intracellulære prosesser og til og med cellelevedyktighet 19, og slik at det er nødvendig å etablere en balanse mellom effektiv bleking og minimal fototoksisitet, og videre, bør celleviabilitet alltid kontrolleres etter live-cell imaging-eksperimenter. Gitt den korte innspillingstiden, gjorde vi ikke vurdere gentoksiske effekten av kort bølgelengde lys (405 nm) i eksempelet som er beskrevet i denne artikkelen, men hvis en lengre eksperiment er nødvendig, en 405 nm laser linje skal ikke brukes.
Vi valgte å ikke bruke en samsvarende tilnærming til transmisjonselektronmikroskopi på grunn av den heterogene natur OSER arkitektur og det faktum at vi ønsket å observere så mange celler (og strukturer) som mulig. Mangfoldet av den fine strukturen av protein aggregater i cellene kan være en viktig funksjon av forskjellige sykdommer, og vi var interessert i å få et bredt spekter av SAmples, mens en samsvarende tilnærming tillater observasjon av færre hendelser i løpet av samme periode. Imidlertid bør trene seg lys-elektronmikroskopi (CLEM) være førstevalget når undersøke hendelser i strukturer som ikke kan lett identifiseres (for eksempel mindre fremtredende ER underdomener) eller i et begrenset antall celler (for eksempel mikro-injisert celler). Det er verdt å merke seg at våre eksperimenter ble karakterisert ved en høy grad av effektivitet transfeksjon (minst 30% av cellene ble transfektert), også mulighet for å observere OSER strukturer noncorrelatively er ganske begrenset.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne har ingenting å avsløre.
Acknowledgments
Forfatterne er takknemlige for Fondazione Filarete for sin hjelp og støtte i publikasjonen av denne artikkelen. Vi vil også gjerne takke Centro Europeo di Nanomedicina for bruk av Tecnai G2 transmisjonselektronmikroskop.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444-456 (2001).
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H. Toxic proteins in neurodegenerative disease. Science. 296, 1991-1995 (2002).
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}