

Summary
Se describen los métodos de formación de imágenes que utilizamos para investigar la distribución y la movilidad de las proteínas fluorescentes transfectadas residentes en el retículo endoplásmico (RE) por medio de la imagen confocal de células vivas. También analizamos ultraestructuralmente el efecto de su expresión en la arquitectura de este compartimento subcelular.
Abstract
Los lípidos y proteínas en células eucariotas se intercambian continuamente entre los compartimentos celulares, a pesar de que estos conservan su composición y funciones distintivo a pesar de la intensa tráfico interorganelle molecular. Las técnicas descritas en este documento son poderosos medios de estudiar las proteínas y la movilidad de los lípidos y el tráfico in vivo y en su entorno fisiológico. La recuperación de la fluorescencia después de photobleaching (FRAP) y la pérdida de fluorescencia en photobleaching (FLIP) se utilizan ampliamente técnicas de formación de imágenes en vivo de células para el estudio de tráfico intracelular a través de la vía endocítica-exo, la continuidad entre orgánulos o subcompartimentos, la formación de complejos de proteínas, y la proteína de localización en microdominios lipídicos, todos los cuales se pueden observar en condiciones fisiológicas y patológicas. Las limitaciones de estos enfoques se deben principalmente a la utilización de las proteínas de fusión fluorescentes, y sus posibles desventajas incluyen artefactos sobreexpresande iones en las células y la posibilidad de diferencias en el plegamiento y la localización de las proteínas etiquetadas y nativas. Por último, como el límite de resolución de la microscopía óptica (aproximadamente 200 nm) no permitir la investigación de la estructura fina de la sala de emergencias o los subcompartimentos específicos que puede originarse en las células bajo estrés (es decir, la hipoxia, la administración del fármaco, la expresión sobre-de transmembrana proteínas residentes ER) o bajo condiciones patológicas, combinamos imágenes de células vivas de las células transfectadas cultivadas con ultraestructural análisis basado en la microscopía electrónica de transmisión.
Introduction
El descubrimiento de la proteína verde fluorescente (GFP) y sus variantes espectrales, y el desarrollo paralelo de la microscopía de fluorescencia, se han abierto por completo nuevas avenidas para la investigación del comportamiento de proteínas en las células. Técnicas tales como la recuperación de la fluorescencia después de photobleaching (FRAP) y la pérdida de fluorescencia en photobleaching (FLIP), que son posibles debido a la capacidad intrínseca de fluoróforos para extinguir su fluorescencia bajo iluminación intensa, se basan en imágenes de células vivas confocal y el uso de transfectaron proteínas de fusión fluorescente 1-3. Son ampliamente utilizados para evaluar no sólo la localización de las proteínas, sino también su movilidad y el transporte vesicular, que puede revelar pistas importantes en cuanto a su función 4.
La característica única de las células eucariotas es la presencia de compartimentos intracelulares que tienen composiciones de lípidos y de proteínas específicas. Aunque orgánulos son físicamente isolated, que necesitan comunicarse unos con otros y compartir componentes moleculares con el fin de mantener la homeostasis celular. La vía secretora garantiza que las proteínas y lípidos sintetizados en el ER llegar al destino final correcto en el que ejercen su función. Orgánulos intracelulares también se pueden conectar por los sitios de contacto dinámicos que permiten a las moléculas (lípidos) para ser intercambiados directamente entre compartimentos. Además, muchas proteínas tienen que reunidos en grandes complejos heteroméricos o asociada a determinadas especies de lípidos (balsas lipídicas / microdominios) para llegar a ser funcionalmente activo o ser transportado a su destino final. Todos estos aspectos biológicos influyen en gran medida las propiedades cinéticas de las proteínas, y por lo tanto puede ser investigado apropiadamente por medio de las técnicas descritas a continuación.
Nuestro grupo ha utilizado ampliamente FRAP y FLIP combinado con microscopia electrónica para estudiar la arquitectura de la sala de emergencia y de sus subsistemasdominios. El ER es la primera estación de la vía secretora y juega un papel clave en proteínas y lípidos de clasificación 5. Es un orgánulo muy dinámico cuya subdominios distintos reflejar sus muchas funciones diferentes (es decir, las proteínas y la biosíntesis de lípidos y la trata de personas, plegamiento de proteínas, Ca 2 + de almacenamiento y liberación, y el metabolismo de xenobióticos). Sin embargo, a pesar de que son morfológicamente, espacialmente, y funcionalmente distintos, estos dominios son continuas entre sí, y su abundancia relativa se pueden modificar en las células bajo condiciones fisiológicas y patológicas. Los más conocidos y dominios generalmente espacialmente segregados de la sala de emergencias son la envoltura nuclear, y el RE liso y áspera, sin embargo, nosotros y otros han demostrado que hay estructuras ER con una arquitectura más elaborada y la organización tridimensional en varios tipos de células y tejidos en condiciones fisiológicas que también pueden ser inducidos por medio de estímulos estresantes tales como la hipoxia, la drogala administración, o la sobre-expresión de proteínas transmembrana-ER residente 2,6 (y las referencias en él).
Recientemente, también hemos demostrado la presencia de tales estructuras en los modelos celulares de enfermedades humanas 1,7. Procedentes de las cisternas apiladas de RE liso, se les dio el nombre colectivo de retículo endoplásmico liso organizada (RPEO) en 2003 6, a pesar de que también se conocen como karmellae, laminillas y ER cristaloide sobre la base de su arquitectura, que, al igual que su tamaño, puede variar. Después de que las células son transfectadas con GFP fusionado a la región citosólica de cola-anclado (TA) proteínas ER-residente (d EGFP-ER), la tendencia débilmente dimerizing de GFP en trans altera dramáticamente la organización y estructura de la sala de emergencias. FRAP y FLIP experimentos mostraron que d EGFP-ER es libre de difundirse dentro de RPEO, y el hecho de que se mueve desde el ER reticular a la RPEO y viceversa </ Em>, indica que los agregados son continuas con la ER reticular circundante. El análisis ultraestructural nos ha permitido correlacionar los datos de fluorescencia con una descripción detallada de la arquitectura RPEO y organización a nivel de nanoescala: RPEO siempre están compuestos de pilas de cisternas emparejado del RE liso pero pueden tener diferentes formas de organización espacial, como dispuestos regularmente sinusoidal matrices o espirales o matrices tubulares "cristaloides" hexagonales. Estos reordenamientos conducen a morfologías cúbicos 8 que, ya que se han encontrado en las células bajo condiciones fisiológicas 9 y siguientes tensiones tales como la hipoxia 10, el tratamiento de drogas 11, y el cáncer 9, pueden tener un potencial significativo como marcadores ultraestructurales.
Después de esta primera demostración del uso de proteínas de fusión GFP, utilizamos los experimentos de imagen para analizar la proliferación de dominios ER en respuesta a los tratamientos farmacológicos 12, assess la tendencia de las proteínas fluorescentes para oligomerise en las células 13, y para investigar el papel de un mutante, ALS ligado a proteína TA en la formación de agregados intracelulares de origen ER que pueden ser relevantes para su 1,8 patogenicidad. Se ha sugerido que la formación de agregados intracelulares (que se produce en muchas enfermedades neurodegenerativas 14) puede ser un mecanismo protector diseñado para evitar las interacciones entre las proteínas mutantes tóxicos y los componentes de las células circundantes 15.
Lo que sigue es una descripción de una combinación de métodos de microscopía óptica y electrónica para la investigación de las construcciones cuyo C-terminal de dominios hidrofóbicos se insertan en la membrana del ER, y un análisis de su comportamiento dinámico y los efectos de su sobre-expresión de dominio ER arquitectura en células cultivadas (véase la Figura 1 para un diagrama de flujo del protocolo experimental).
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. El plásmido, cultivo celular, y la transfección con ER proteínas fluorescentes
- El plásmido usado en este estudio consiste en una versión mejorada de GFP fusionada en su extremo C-terminal a la región de la cola de la isoforma ER de rata citocromo b (5) (abreviado aquí como B (5)) a través de una secuencia de engarce. La región de la cola contiene toda la secuencia (Pro94-Asp134) que permanece asociado a la membrana después de la escisión de tripsina de B nativa (5), incluyendo el TMD 17-residuo (dominio transmembrana), flanqueado por secuencias polares aguas arriba y aguas abajo (UPS y DPS) . El enlazador consiste en el epítopo myc seguido de [(Gly) 4 Ser] 3, y todo el ADNc se insertó en los sitios Hind3-Xba1 del vector de expresión de mamíferos pCDNA3. Los detalles de la construcción de este plásmido se han descrito en una publicación anterior en la que se conoce como GFP-ER 16.
- Crecer células COS-7 en medio Eagle modificado de Dulbecco (DMEM) suplementado con 10% de queso Fetal de suero bovino, 2 mM de L-glutamina, 1% de penicilina / estreptomicina en una incubadora a 37 ° C y 10% de CO 2.
- La transfección. Placa 3 x 10 5 células sobre un cubreobjetos de vidrio redonda en una placa de 6 pocillos y, al día siguiente, transfectar con el sistema JetPEI como se describe por el fabricante. Tenga en cuenta que la relación JetPEI / ADN óptima ha sido probado con el fin de establecer la máxima eficiencia de la transfección dependiendo de la plásmido y la línea celular utilizada: en nuestro caso, un JetPEI: relación de ADN de 2:01 conduce a 70-80% de eficiencia de transfección.
2. Vivo de fluorescencia confocal de barrido Microscopía
- Imágenes de células vivas. Coloque el cubreobjetos en la que las células transfectadas se sembraron en una cámara de cultivo celular de acero para cubreobjetos 24 mm relleno con DMEM w / o rojo de fenol, suplementado con 10% de FBS, 2 mM de L-glutamina, 1% penicilina / estreptomicina, 25 mM de HEPES , 50 mg / ml de cicloheximida y 1:100 OxyFluor para evitar que las muestras de photobleaching. Un microscópicas confocal SP5e equipado con una incubadora de CO2 a temperatura controlada (37 º C y 5% CO 2) se utiliza para experimentos de imagen de células vivas, con d GFP-ER se visualizó usando un láser de 488 nm y un filtro de 525/50 emisión del paso de banda.
- Recuperación de fluorescencia después de photobleaching (FRAP). Dibujar una región de interés (ROI), correspondiente a una estructura Oser y lejía usando 20 iteraciones y una combinación de 488 nm (100% de un láser de 30 mW de argón, correspondiente a 5.5-6 mW en la muestra) y 405 nm (60% de una 30 mW diodo láser de 405, lo que corresponde a 11,6 mW en la muestra) láseres que, en nuestra experiencia, conduce a fotoblanqueo eficiente y rápida.
- Registre la recuperación de la fluorescencia en las regiones de interés blanqueados mediante la adopción de un único fotograma cada 10 segundos durante 10 minutos (tiempo de píxel = 1,61 microsegundos / px).
- Pérdida de fluorescencia en photobleaching (FLIP). Dibuje un retorno de la inversión correspondiente a una estructura RPEO, y blanqueador como se describió anteriormente. El blanqueo se repite cada 30 segundos,y las imágenes post-blanqueamiento se registran cada 10 segundos durante 30 minutos (tiempo de píxel = 1,61 microsegundos / px).
- FRAP y el análisis de la FLIP. Todas las imágenes son analizadas utilizando software ImageJ ( http://rsbweb.nih.gov/ij/download.html ). En los experimentos FRAP, la recuperación de la fluorescencia de la ROI blanqueada se mide con el tiempo y se normalizaron a la fluorescencia total de células blanqueada, que siempre se comprueba para ser constante en el tiempo.
- Para los experimentos de FLIP, dibuje un retorno de la inversión fuera de los RPEO blanqueada y cubriendo toda la célula. Medir su intensidad de fluorescencia con el tiempo y normalizar para los niveles de fluorescencia de un retorno de la inversión dibujadas en una célula sin blanquear con el fin de corregir para cualquier disminución de la fluorescencia causada por la propia formación de imágenes.
- En todos los experimentos, se resta la señal de fondo (determinado en un área fuera de las células) de las intensidades fluorescentes de las regiones de interés. Por último, trazar los resultados utilizando Grsoftware aphPad Prism.
3. Análisis ultraestructural por Medio de Microscopía Electrónica de Transmisión
Dada la toxicidad de muchos de los reactivos, todos los procedimientos deben llevarse a cabo usando un laboratorio apropiado. abrigo y los guantes bajo una campana de humos.
- Después de retirar el cubreobjetos de la placa de Petri, fijar las células restantes cultivadas en la parte inferior del plato como una monocapa usando filtrada 2% de glutaraldehído en 0,1 M de tampón de cacodilato, pH 7,4, durante 10 min a temperatura ambiente.
- Raspar las células utilizando un raspador de teflón y transferirlos a 1,5 ml de tubos de Eppendorf. Sedimentar las células por medio de centrifugación a 9000 g durante 10 min. Eliminar el sobrenadante, añadir fijador fresco, y dejar toda la noche a 4 ° C.
- Lavar los pellets con el tampón, a continuación, después de fijar con una solución de 1% de tetróxido de osmio en tampón cacodilato durante 1 hora a temperatura ambiente.
- Enjuague con agua Milli-Q, y en la mancha bloque con1% de acetato de uranilo en agua destilada durante entre 20-60 min.
- Deshidratar las muestras en el aumento de serie de etanol (70%, 80%, 90%, 100%, y 100% durante 10 min cada uno), y se lava brevemente dos veces en óxido de propileno (15 min cada uno).
- Infiltrarse en las muestras en una mezcla de óxido de propileno + Epon (01:01) (de 2 horas para la noche).
- Insertar en resina epoxi Epon curado a 60 ° C durante al menos 24 horas.
- Sección los bloques de resina recortadas manualmente utilizando un ultramicrotomo LEICA UC6 equipado con un cuchillo de diamante 45 ° para obtener secciones con un espesor de 60-70 nm. Recoge las secciones sobre rejillas de cobre de malla 300.
- Teñir las secciones de la red con una solución saturada de acetato de uranilo (20 min) y citrato de plomo (7 min), lavar bien las rejillas sumergiéndolas en agua filtrada bidestilada, y déjelos secar a temperatura ambiente.
- Las rejillas teñidos se observan con un microscopio electrónico de transmisión Tecnai G2, y las imágenes se capturaron con una bottcámara CCD montada om-con diferentes ampliaciones finales (generalmente van de 6,000-39,000 X).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
La Figura 2 muestra un estudio FRAP ejemplo de la movilidad de la proteína. La movilidad de la proteína EGFP D-ER se demuestra por la recuperación de la fluorescencia después de photobleaching rápida en RPEO blanqueados. Para el análisis cuantitativo, la media hora y la fracción celular se obtuvieron a partir de los datos medidos experimentalmente mediante el ajuste de la siguiente ecuación monoexponencial:
F (t) = F + mensaje (post F rec-F) (1-e-t / τ)
donde después F es la señal de fluorescencia después de photobleaching, F rec es el máximo valor de recuperación de fluorescencia que se alcanza después del blanqueo, t el tiempo de registro y τ la constante de tiempo.
Por favor, tenga en cuenta la importancia de la adquisición de imágenes sin píxeles saturadas que puedan alterar la recuperación de fluorescencia y, por consiguiente, el análisis de la movilidad de la proteína. Es is también esencial para normalizar siempre la señal de fluorescencia en el ROI blanqueada a la fluorescencia total de la misma celda con el fin de tener en cuenta las variaciones de intensidad de fluorescencia debido a la decoloración durante la adquisición de la imagen o de los pequeños cambios en el plano de enfoque.
Un ejemplo de un experimento de la FLIP para estudiar la continuidad entre los compartimentos intracelulares se muestra en la Figura 3. RPEO están conectados físicamente con el resto de la sala de emergencia como se demuestra por el vaciado progresivo de la sala de emergencias cuando el dominio RPEO se blanquea de forma continua.
Para un análisis adecuado, la adquisición de píxeles saturados debe ser evitado (véase más arriba) y, además, los parámetros de adquisición se deben configurar con potencias de láser tan bajas como sea posible a fin de evitar photobleaching debido a la adquisición de la imagen. Por esta razón se recomienda encarecidamente a la imagen una célula sin blanquear en el mismo campo que se utiliza para normalizar la señal de fluorescencia de la c blanqueadaell.
Todos los experimentos se tienen que realizar en presencia de cicloheximida, un inhibidor de la traducción, con el fin de evitar cualquier aumento en la señal de fluorescencia ER (y en consecuencia la fluorescencia total) debido a la biosíntesis de proteínas.
Microscopía electrónica de transmisión demostró que los agregados fluorescentes observados en células cultivadas transfectadas con EGFP D-ER representan parches de suave y aplanada cisternas del RE que espacialmente ellos mismos organizado en geometrías 3D bien definidos clasificadas sobre la base de sus patrones: pilas lineales o curvas (a menudo asociado con la envoltura nuclear, que no se muestra) (Figuras 4A y B) que puede ser continua con las regiones de ER sinusoidal (Figura 4A); las membranas en algunas regiones se organizan en celosías con una simetría cuadrada o hexagonal (ER cristaloide, no mostrado ). Cisternas adyacentes están separadas por una fina capa de un poco de electronescitoplasma denso alrededor de 11 nm de espesor que es continua con el citoplasma que rodea a los agregados.

Figura 1. Diagrama de flujo del procedimiento experimental. Las células cultivadas se transfectaron primero con jetPEI (véase el Protocolo) con el fin de sobre-expresar la proteína de fusión fluorescente de interés. Después de 24 horas, las células transfectadas en vivo se visualizan y FRAP y FLIP experimentos se realizaron con un microscopio confocal equipado con una temperatura controlada de CO 2 incubadora, y las imágenes grabadas se exportan y se analizaron con el software correspondiente (por ejemplo, ImageJ). Para el análisis ultraestructural, de las células transfectadas son fijos, sedimentaron y embebidos en Epon bloques de resina epoxi. Ultrathin secciones se obtienen utilizando un cuchillo de diamante, recogida en cOpper rejillas, y se observan bajo un microscopio electrónico de transmisión. Haz clic aquí para ver más grande la figura .

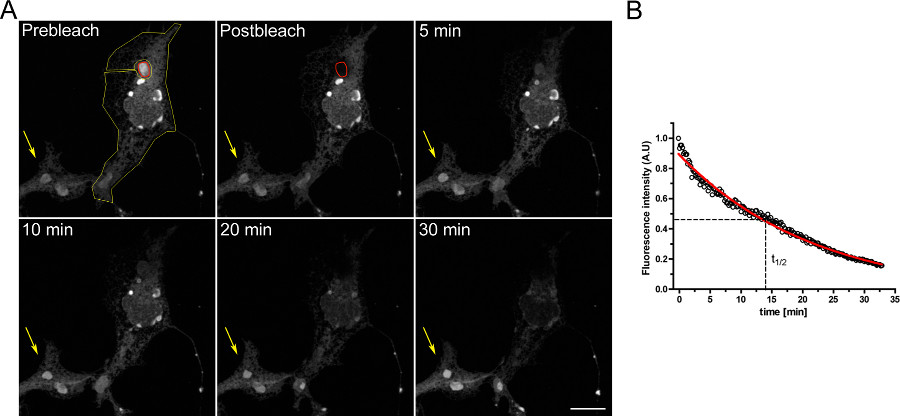
Figura 2. FRAP experimento utilizando células COS-7 transfectadas transitoriamente con d EGFP-ER. A) Dos estructuras RPEO (Rois rojas) fueron blanqueadas, y la recuperación de la fluorescencia se registró en el tiempo. Recuperación de fluorescencia Clear se puede detectar 1 min post-blanqueo, y la señal de más aumentos de 4 minutos más tarde (barra de escala 10 micras) B):. Análisis cuantitativo del experimento FRAP que muestra la media de tiempo de recuperación y la fracción móvil de la d EGFP ER-proteína. Click aquí para ver la figura más grande

Figura 3. FLIP experimento utilizando células COS-7 transfectadas transitoriamente con d EGFP-ER. A) El blanqueo continuo de un RPEO (indicado por el retorno de la inversión de color rojo) provoca una disminución progresiva en la fluorescencia en el resto de la sala de emergencia y en otras estructuras RPEO dentro de la misma célula (indicado por el retorno de la inversión de color amarillo). La flecha amarilla indica una porción de una célula sin blanquear en el que la señal de fluorescencia es constante en el tiempo. (Barra de escala 10 micras). B) Análisis cuantitativo del experimento FLIP. Haga clic aquí para ver más grande la figura
Figura 4. Después de la fijación y la incrustación, se observaron células que expresan altos niveles de D EGFP-ER en el que las estructuras de los RPEO podrían ser detectados por medio de microscopía óptica de fluorescencia a través de un microscopio electrónico de transmisión. A) Vista de baja magnificación de una porción de citoplasma de una célula que contiene un RPEO que consiste en cisternas apiladas y membranas sinusoidales ondulantes. Las mitocondrias (M) se puede ver agrupados en torno a las estructuras de los RPEO, mientras que los ribosomas sólo decore las membranas de las cisternas más externa (puntas de flecha y el recuadro). El espacio nm de espesor 11 electrones de alta densidad entre las membranas es continua con el citoplasma (flecha y cuadro) (L = lisosomas / (auto-) fagosomas) (barra de escala 1,5 m; inserción 0,25 m). B) Un RPEO se puede formar por lamelar ER: es decir, pilas de cisternas del RE aplanada que puede ser continua o fragmented en su aparición en secciones delgadas. Las vesículas en ciernes de las cisternas más externa de la pila de vez en cuando se pueden observar (asterisco) (PM, membrana plasmática) (barra de escala a 150 nm).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Los protocolos y métodos de formación de imágenes descritos en el presente documento se han utilizado para investigar la distribución y la movilidad de las proteínas fluorescentes TA transfectadas residentes en el ER de las células vivas. También hemos analizado el efecto de la expresión sobre-de estas proteínas en la arquitectura de este compartimiento subcelular por medio de análisis ultraestructurales.
La combinación de imagen confocal de células vivas y microscopía electrónica representa es un medio muy poderoso de la investigación de las propiedades dinámicas de las proteínas, y puede proporcionar información importante acerca de la función de proteínas. Los métodos descritos no son mucho tiempo (típicamente tres días de trabajo), y el desarrollo de muchas aplicaciones de software fácil de usar para la adquisición y análisis de imágenes basado hace photobleaching-, imágenes de células vivas relativamente simple.
La principal limitación de estas técnicas es el uso de proteínas de fusión fluorescente debido a que laetiqueta fluorescente puede afectar el plegamiento y / o montaje adecuado de la proteína de interés. Además, la sobre expresión puede alterar el comportamiento de las proteínas transfectadas, marcado con fluorescencia, y por lo tanto no pueden reflejar las propiedades reales de proteínas endógenas, sin embargo, esto se puede superar mediante el uso de las células inducibles y transfectadas de manera estable en la que el nivel de expresión puede ser precisamente modulada obtener niveles comparables con los de la proteína endógena 1,7. La tendencia de los MF a oligomerise ha sido ampliamente documentada y podría alterar de manera significativa el comportamiento (es decir, la cinética, las interacciones proteína-proteína no deseados y la formación de agregados) de las proteínas quiméricas. Por consiguiente, el uso de proteínas fluorescentes monoméricas optimizados se debe considerar 17.
Otro aspecto crítico de los estudios de formación de imágenes dinámicas utilizando fluorescencia y fotoblanqueo es el tiempo necesario para blanquear la fluorescencia eficiente y medir la fluorescencia de rerecuperación (y por lo tanto la movilidad de la proteína), precisamente, que también depende del área de la ROI y espesor celular local. Si una proteína GFP-etiquetados dado tiene un alto coeficiente de difusión, la difusión puede ocurrir durante el blanqueo y por lo tanto interferir con las mediciones de tiempo de recuperación. Con el fin de obtener el blanqueamiento rápido y eficaz, se recomienda encarecidamente que un "Zoom in" de función (si está disponible) y más de una línea de láser se utilizan. Aunque el uso de un módulo de exploración rápida (es decir, un escáner de resonancia) puede mejorar en gran medida la velocidad de formación de imágenes durante la fase de recuperación de un experimento, en nuestras manos sino que también reduce considerablemente la eficiencia de blanqueo. Sin embargo, los sistemas alternativos de escaneado (tales como un disco giratorio equipado con un dispositivo de fotoblanqueo dedicado), y los láseres más potentes pueden mejorar tanto la eficiencia de blanqueo y la velocidad de adquisición.
La mayoría de las proteínas fluorescentes utilizados en FRAP y FLIP experimentos muestran algún grado de fotoblanqueo reversible y bliNking que deben tenerse en cuenta al realizar análisis cuantitativos. Las fluctuaciones entre estados fluorescentes y oscuras se producen en el segundo lugar a escala temporal minutos. Para EGFP, se ha demostrado que durante los experimentos de blanqueo, las variaciones de fluorescencia pueden implican menos de 10% de las moléculas, por lo tanto en el presente protocolo de este fenómeno es insignificante. Si todas las condiciones se mantienen constantes, esto va a introducir un sesgo constante en los resultados. Si se utilizan otras proteínas fluorescentes, en el que la fracción reversible es significativamente más alta (es decir, YFP), o para detectar y evaluar la reversibilidad photobleaching, esto se puede hacer mediante la medición de la recuperación después de fluorescencia photobleaching en toda la célula viva; si se observa la recuperación esto puede sólo ser el resultado de photobleaching reversibilidad 18.
La toxicidad potencial de la luz durante los experimentos es otro factor crítico, sobre todo porque fotoblanqueo requiere iluminación intensa. Es well sabe que fluoróforos excitados tienden a reaccionar con el oxígeno para producir radicales libres que pueden afectar a diferentes procesos intracelulares e incluso la viabilidad celular 19, por lo que es necesario establecer un equilibrio entre el blanqueamiento eficiente y fototoxicidad mínima y, además, la viabilidad celular siempre se debe revisar después de los experimentos de imagen de células vivas. Teniendo en cuenta el tiempo de grabación corto, no consideramos el efecto genotóxico de luz de longitud de onda corta (405 nm) en el ejemplo descrito en este artículo, pero, si se necesita un experimento más largo, una línea láser 405 nm no deberían utilizarse.
Decidimos no utilizar un enfoque correlativo a la microscopía electrónica de transmisión debido a la naturaleza heterogénea de la arquitectura RPEO y el hecho de que queríamos para observar la mayor cantidad de células (y estructuras) como sea posible. La diversidad de la estructura fina de los agregados de proteínas en las células puede ser una característica clave de las diferentes enfermedades y estábamos interesados en obtener una amplia gama de samples, mientras que un enfoque correlativo permite la observación de un menor número de eventos durante el mismo período de tiempo. Sin embargo, el microscopio óptico-electrónica correlativa (CLEM) debe ser la primera opción en la investigación de los acontecimientos en estructuras que no pueden ser identificados con facilidad (como subdominios ER menos prominentes) o en un número limitado de células (como las células micro-inyectada). Vale la pena señalar que nuestros experimentos se caracterizan por un alto grado de eficacia de la transfección (por lo menos 30% de las células fueron transfectadas), de lo contrario la posibilidad de observar estructuras RPEO noncorrelatively es bastante limitada.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores no tienen nada que revelar.
Acknowledgments
Los autores agradecen a la Fundación Filarete por su ayuda y apoyo en la publicación de este artículo. También nos gustaría agradecer Centro Europeo di nanomedicina para el uso del microscopio electrónico de transmisión Tecnai G2.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444-456 (2001).
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H. Toxic proteins in neurodegenerative disease. Science. 296, 1991-1995 (2002).
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}