Microscopy
Microscopy samples can and should be analyzed immediately to ensure they are of the desired quality. To measure abundance and size distribution of the bacterial community, DAPI slides will be examined by epifluorescence microscopy (excitation/emission: 358/461 nm, see McDole et al. 2012) (Figure 6A). Cell counts and dimensions can be collected using imaging software (e.g., ImagePro or ImageJ). From length and width measurements cell volumes (V) are derived by making the assumption that all cells have the shape of cylinders with hemispherical caps using the following equation:
V = π /4 × w2 (L –w/3)
where L is length and w is width of each cell23,4. Microbial biomass can then be estimated using previously established size-dependent relationships for marine microbial communities24. Generally marine bacteria range in length from 0.1-4 µm, but go up to ~ 8 µm in some locations.
While the filters (0.2 µm) stained with DAPI will only show bacteria, the filters used for the SYBR Gold stain (0.02 µm) contain bacteria and viruses. Measuring the abundance of viruses follows the same protocol as for microbes, however an excitation of 325-375 nm will be used and the emission maximum is at 537 nm (Figure 6B).
In order to generate quantitative data the sampling volume may need to be adjusted depending on the viral abundance in the original sample. The correct volume to filter is best determined empirically for a given body of water. Examples of micrographs containing samples with varying viral concentrations are illustrated in Figure 7.
Results from previous studies suggest that Virus to Microbe ratios (VMR) generally range from 1 to 50 in aquatic systems25-29, and in between 3 and 20, with an average of approximately 6 in coral reef systems (Knowles unpublished data).
Flow Cytometry
In addition to the direct counts and size estimation of the microbial community, assessments of the ratio of autotrophic to heterotrophic microbes via flow cytometry can further be extracted from the collected samples (exemplary flow cytometry output given in Figure 8). To determine the number of total Bacteria cells, samples are stained with SYBR Green I and a photomultiplier tube with a 530/30 bandpass filter is used for detection. A channel for chlorophyll (back to back LP mirrors resulting in a range of 675-735 nm) and phycoerytherin (585/42 bandpass filter) is used to count the abundance of autotrophs in unstained samples. To determine the abundance of heterotrophic microbes, the autotrophic counts from the non-stained portion are then subtracted from the SYBR-stained total count (McDole et al. submitted).
Viral Metagenomics
Viral metagenomics utilizes a culture-independent molecular approach to viral ecology, using total genomic sequence information to determine community structure and function. Metagenomics has been advancing our understanding of the complexity and diversity of viral communities at local17 to global scales30. The recent development of a wide array of bioinformatic tools for analysis of viral metagenomic data has decreased computational bottlenecks, allowing for a more comprehensive view the of the virosphere through the lens of metagenomics.
The methods presented here highlight the isolation and enrichment of viral particles from seawater using a combination of pre-filtration with large pore size nylon mesh to remove debris and cellular material, concentration of samples with TFF (Figure 3) and a subsequent 0.45 μm filtration to remove larger cells. This method produces roughly a 100x concentrated sample (Figures 5B-5E) which allows us to perform downstream processes in smaller volumes. In order to prevent growth of any remaining microbial cells in the viral lysate and therefore changes in the viral abundance of the sample during the long transit time between field station and laboratory, chloroform is often added to a final concentration of 2% for storage. Following isolation and purification of VLPs, epifluorescence microscopy with nucleic acid dyes such as SYBR Gold are used to verify the presence and purity of the viral particles (Figures 5B-5E).
Here, we present two viral metagenomes from the Southern Line Island coral reef, specifically, the Starbuck (Star7) and Millennium (CAR9; previously known as Caroline) islands. A total volume of 120 L sample water was collected from each site at a depth of 10 meters and processed as described in section 3.7 and 5.2. The VLPs purified from the cesium chloride gradient centrifugation were 3.3 x 108 particles/ml for CAR9 (Figure 5D) and 2.9 x 109 particles per ml for Star7 as verified according to the method described in paragraph 4 (Figure 5B). The total amount of DNA isolated from these samples were around 400 ng based on the Nanodrop measurement. DNA was amplified using Phi29 polymerase and sequenced by the Environmental Genomics Core facility in 2011. The characteristics of the sequence data is presented in Table 1. Based on similarity searches using existing databases, the majority (>70%) of the sequences often ended up uncharacterized, i.e., unknown origins and functions. Similarly, the two viromes presented here presented more than 97% of unknown sequences. As a result, non-database dependent analysis was used for analysis and opened up a new arm of research opportunity on the “dark matter” in viral metagenomics (Seguritan et al. in press).
Microbial Metagenomics
The analysis of microbial metagenomes allows for the characterization of the microbial community present and the functional description of these communities. Examples include estimates of the presence of pathogens and virulence5,22 or comparisons between changes in macrobial community structure or available nutrients and abundance of species and their predominant metabolic pathways (Figure 9; Kelly et al. 2014).
Water Chemistry
Water chemistry parameters generally take extensive analytical processing to generate the desired data; samples for DOC analysis will be measured via high temperature catalytic oxidation31,32, particulate organic matter (POM) via isotope ratio mass spectrometry coupled to an elemental analyzer 33-35, and inorganic nutrients by Flow Injection Analysis36,12,37. After a successful completion of all the analyses, information as shown in Table 1 will be available to complement the characterization of the microbial and viral communities. This information can be complimented with stable isotope ratios of organic carbon and nitrogen samples (See Haas et al.35) and ratios between autotrophs and heterotrophs (McDole et al. submitted).
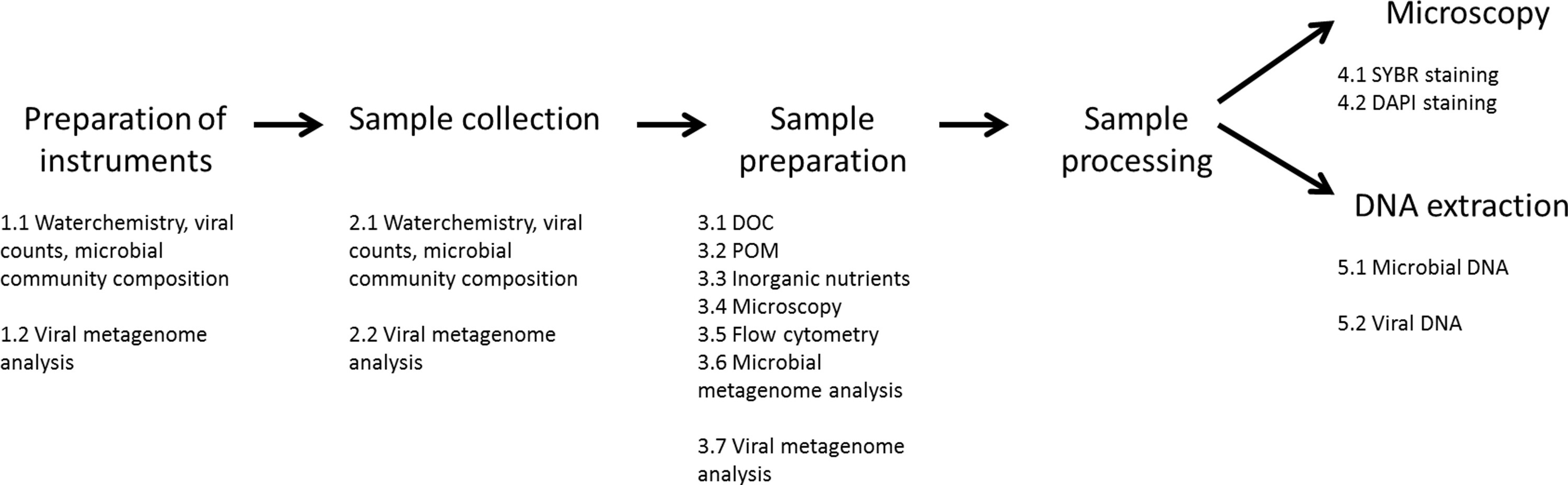
Figure 1. Overview of methods described in the protocol section. Please click here to view a larger version of this figure.
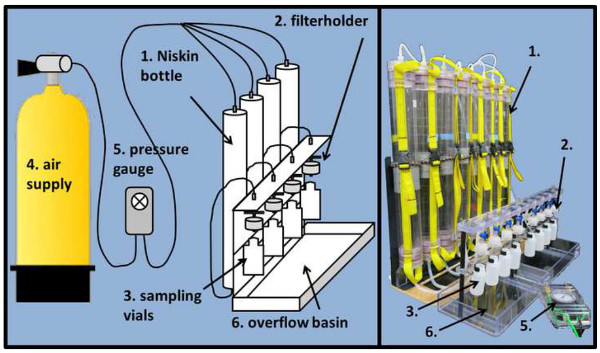
Figure 2. Sample processing rig setup. The filtration setup introduced here (schematic drawing left, actual picture right panel) is not necessarily required to generate samples but will expedite the process significantly. The setup consists of a backplate, on which the Hatay Niskin units (1) are mounted. On the outlet facing downwards, Hatay Niskin units are connected with silicone tubing to the in-line filter holders (2). These let the sampled water pass through the respective filter into the HDPE sampling vials (3) mounted right underneath. The filtration process is accelerated through pressurized air (4), which and regulated by a pressure gage (5). An overflow basin prevents spilling water during the changing of the HDPE vials or POM filtration. Please click here to view a larger version of this figure.

Figure 3. Tangential flow filter lineup (modified from Thurber et al.19). Nylon mesh filtered sample water is pumped from the reservoir (1) via a peristaltic pump (2) to a tangential flow filter (3). The sample water returns to the reservoir (1) along the return line (4) in the absence of backpressure. When backpressure is applied through a hose clamp (5) on the return line, water passes through the filter and is discarded or delivered to the filtrate reservoir (6). Sample water is replaced as it is concentrated out the filtrate line until all the sample water is concentrated in the tubing lines.
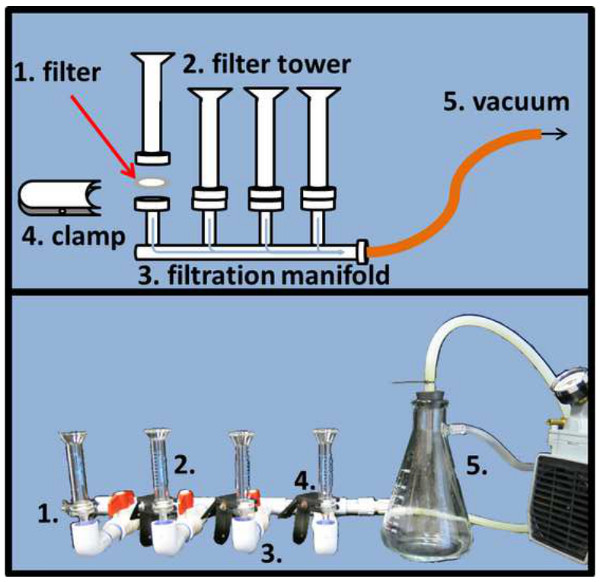
Figure 4. Microscope slide filtration setup. To evenly distribute DAPI and SYBR Gold stained samples on the respective alumina matrix filter (1), filters need to be placed in between the filter tower (2) and stem of the filtration manifold and fixed with a clamp (4). A pressure regulated vacuum (5) will expedite the filtration process (schematic drawing top, actual picture bottom panel). Please click here to view a larger version of this figure.
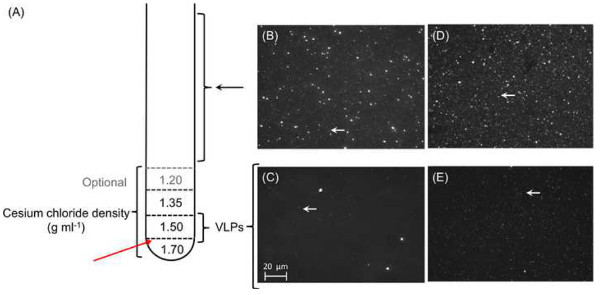
Figure 5. Purification and concentration of phage particles. A total of 1 ml of each cesium chloride density gradient is layered on top of each other prior to leading the pre-treated sample (A). Each layer should be marked outside the tube to facilitate the extraction after ultracentrifugation. Red arrow marks where the tube will be pierced to extract the VLP fraction. (B-E) Micrographs of viral concentrates: Epifluorescence micrographs of 0.45 μm-filtered representative viral concentrates of Star7 (B) and CAR9 (D). Large particles including bacterial cells were visible from the 0.45 μm-filtrates from both Star7 (B) and CAR9 (D) samples, whereas only VLPs (white arrows) remain after cesium chloride gradient ultracentrifugation; Star 7 (C) and CAR9 (E). Please click here to view a larger version of this figure.

Figure 6. Example of DAPI and SYBR Gold micrograph analysis. Screenshot from the analysis of (A) DAPI and (B) SYBR Gold stained slide example. (A) Length and width (µm) of all highlighted particles (= microbes) can be assessed with the help of an imaging program. Note the aggregation in the center of the image. These clusters will need to be excluded from subsequent analysis. (B) During analysis viruses and bacteria need to be binned by size thresholds into VLP and cellular size ranges (VLP red, cellular green). Note that there are usually some faint VLPs uncategorized by the respective imaging program. These VLPs appearing as faint white dots must be manually counted and added to the automated viral counts. Please click here to view a larger version of this figure.

Figure 7. Concentrations of SYBR Gold stained samples. Micrographs of a 0.02 alumina matrix filter containing various numbers of SYBR stained virus samples. Panel A shows a filter containing a suitable amount of seawater filtered, whereas the concentrations on the filter shown in B are too high and in C too low for reliable counts. Please click here to view a larger version of this figure.
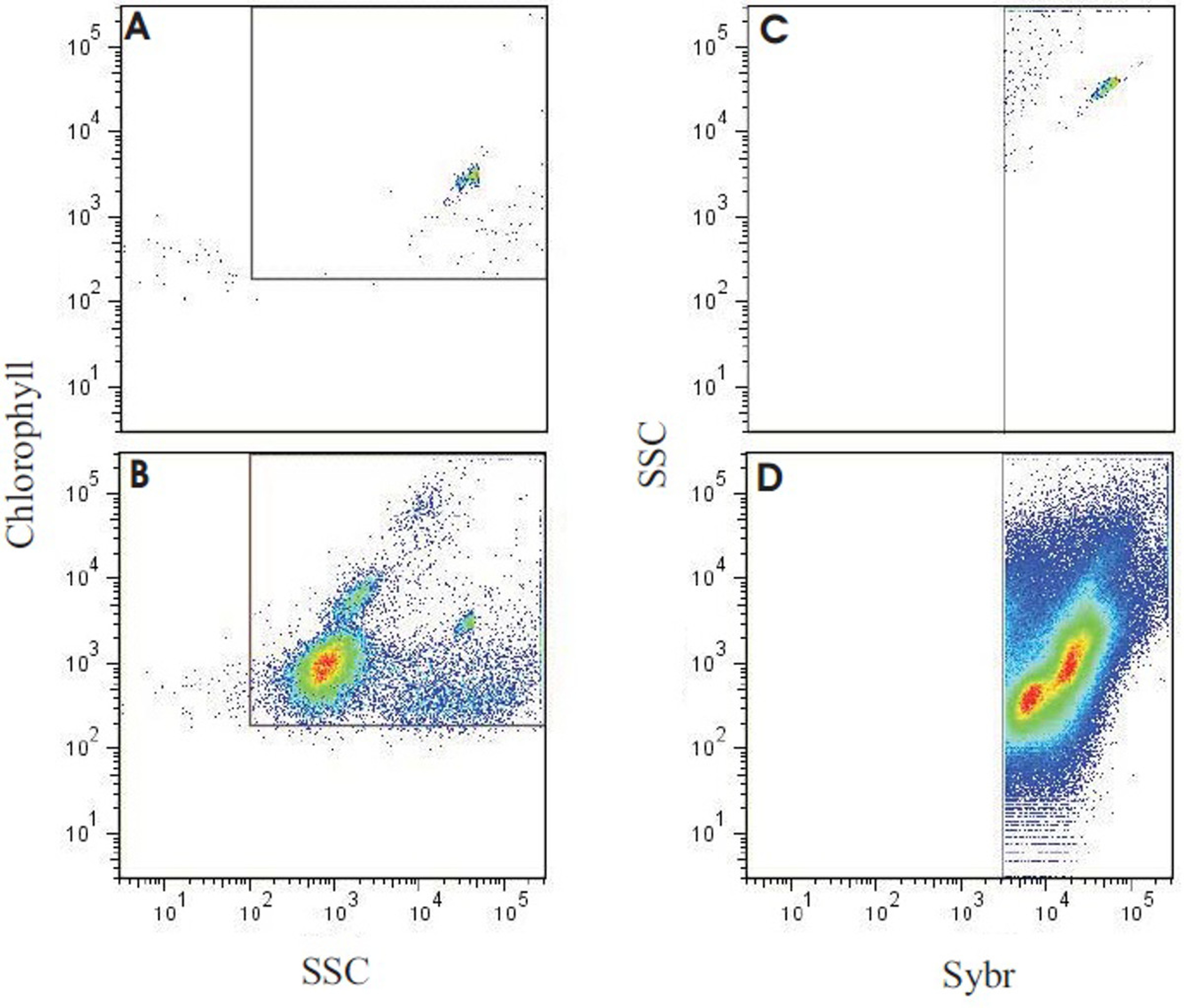
Figure 8. Results of flow cytometry analysis output of a reef-water sample. Panel (A) shows molecular grade water only with yellow-green fluorescent microspheres (0.75 μm), used to verify minimal background with the instrument settings. Panel (B) depicts the output of the reef water sample run with identical settings and layout as in A. Note that the beads can still be seen in the same location, along with several populations of autotrophs. (C) The same sample as in (B), used to back-gate the flow cytometer, targeting SYBR positive events (autotrophic + heterotrophic count) and minimizing background. (D) The representative reef-water sample stained with SYBR Green I. Using the gating established in (C), total microbe counts were generated, and heterotrophic and autotrophic microbes partitioned by chlorophyll autofluorescence (see McDole et al.4). Please click here to view a larger version of this figure.
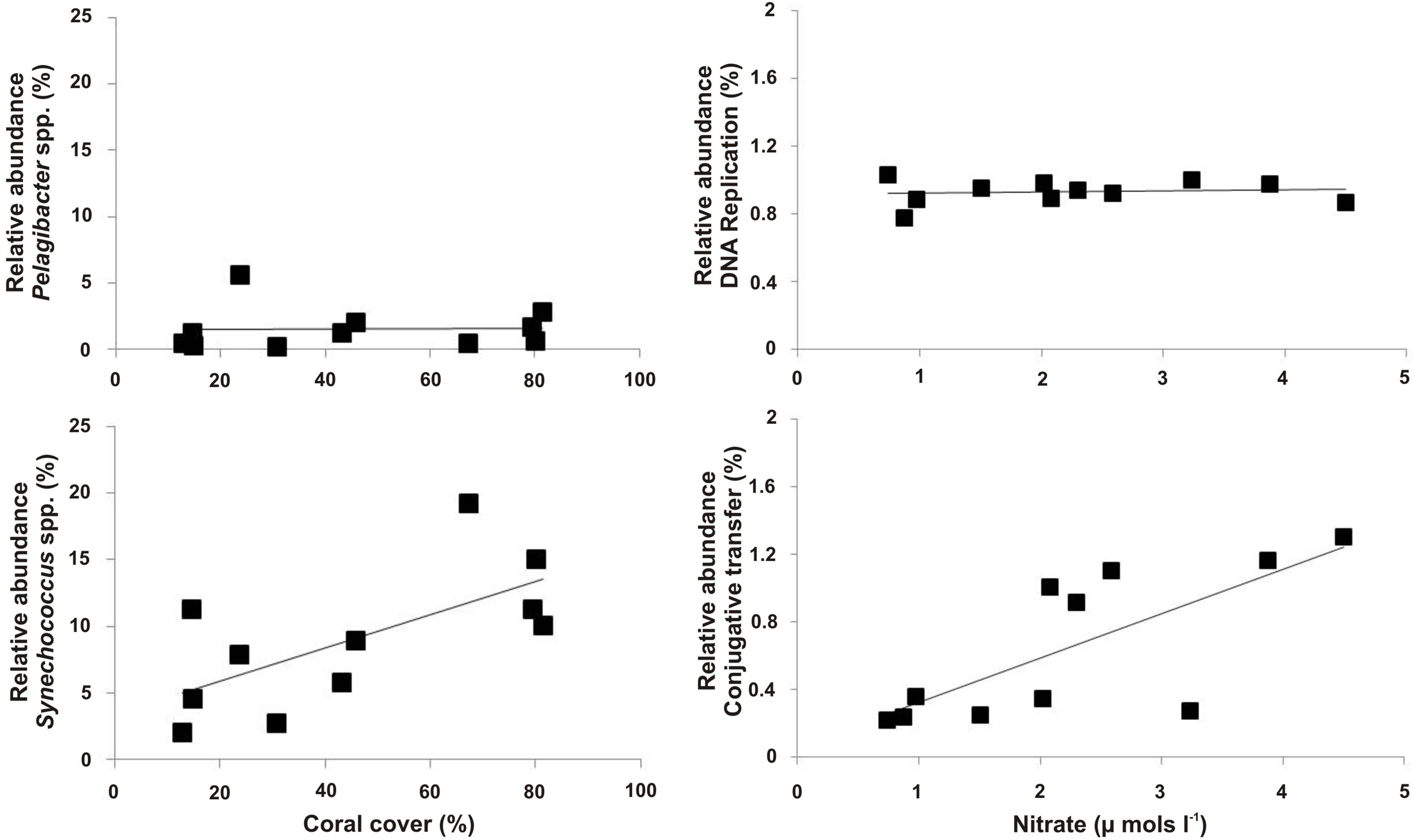
Figure 9. Example of microbial metagenome data. Patterns between the microbial metagenomic sequence data and site dependent variables. Here, the relative abundance of Synechococcus spp. was positively correlated with the percent cover of coral, whereas Pelagibacter spp. was not (left panels). The metabolic pathway for conjugative transfer was positively correlated with nitrate concentrations, while the metabolic pathway for DNA repair was consistent between all metagenomes (right panels). Please click here to view a larger version of this figure.
| Star 7 | CAR9 | |
| Total Number of Reads | 939,311 | 591,600 |
| Number of Preprocessed Reads | 579,664 | 360,246 |
| Mean Sequence Length (bp) | 395 ± 131 | 451 ± 159 |
| Annotated Sequences (%) | 42,218 (7.3%) | 9,117 (2.5%) |
| Unknown (%) | 537,446 (92.7%) | 351,129 (97.5%) |
Table 1. Data characteristics of two viromes generated from the Southern Line Islands, Starbuck and Millennium. The degree of annotation is based on BLAT using the MG-RAST server.

Table 2. Representative results showing data from various sites during one expedition. The here assessed parameters include water chemistry measurements paired with microbial and viral abundances. The table further contains file names referring to the respective metagenomic data for each sampling location. Carbon and inorganic nutrient concentrations are given in μmol/L. Please click here to view a larger version of this table.
