

Summary
Introducing multiple genomic alterations into cyanobacteria is an essential tool in the development of strains for industrial and basic research purposes. We describe a system for generating unmarked mutants in the model cyanobacterial species Synechocystis sp. PCC6803 and marked mutants in Synechococcus sp. PCC7002.
Abstract
Cyanobacteria are ecologically important organisms and potential platforms for production of biofuels and useful industrial products. Genetic manipulation of cyanobacteria, especially model organisms such as Synechocystis sp. PCC6803 and Synechococcus sp. PCC7002, is a key tool for both basic and applied research. Generation of unmarked mutants, whereby chromosomal alterations are introduced into a strain via insertion of an antibiotic resistance cassette (a manipulatable fragment of DNA containing one or more genes), followed by subsequent removal of this cassette using a negative selectable marker, is a particularly powerful technique. Unmarked mutants can be repeatedly genetically manipulated, allowing as many alterations to be introduced into a strain as desired. In addition, the absence of genes encoding antibiotic resistance proteins in the mutated strain is desirable, as it avoids the possibility of 'escape' of antibiotic resistant organisms into the environment. However, detailed methods for repeated rounds of genetic manipulation of cyanobacteria are not well described in the scientific literature. Here we provide a comprehensive description of this technique, which we have successfully used to generate mutants with multiple deletions, single point mutations within a gene of interest and insertion of novel gene cassettes.
Introduction
Cyanobacteria are an evolutionarily ancient and diverse phylum of bacteria found in nearly every natural environment on Earth. In marine ecosystems they are particularly abundant and play a key role in many nutrient cycles, accounting for approximately half of carbon fixation1, the majority of nitrogen fixation2 and hundreds of millions of tons of hydrocarbon production3 in the oceans annually. Chloroplasts, the organelle responsible for photosynthesis in eukaryotic algae and plants, are likely to have evolved from a cyanobacterium that was engulfed by a host organism4. Cyanobacteria have proved useful model organisms for the study of photosynthesis, electron transport5 and biochemical pathways, many of which are conserved in plants. In addition cyanobacteria are increasingly being used for production of food, biofuels6, electricity7 and industrial compounds8, due to their highly efficient conversion of water and CO2 to biomass using solar energy9. Many species can be cultivated on non-arable land with minimal nutrients and seawater, suggesting that cyanobacteria could potentially be grown at large scale without affecting agricultural production. Certain species are also sources of natural products, including antifungal, antibacterial and anti-cancer compounds10,11.
The ability to generate mutants is key to understanding cyanobacterial photosynthesis, biochemistry and physiology, and essential for development of strains for industrial purposes. The majority of published studies generate genetically modified strains by insertion of an antibiotic resistance cassette into the site of interest. This limits the number of mutations that can be introduced into a strain, as only a few antibiotic resistance cassettes are available for use in cyanobacteria. Strains containing genes conferring antibiotic resistance cannot be used for industrial production in open ponds, which is likely to be the only cost-effective means to produce biofuels and other low value products12. The generation of unmarked mutants overcomes these limitations. Unmarked mutants contain no foreign DNA, unless intentionally included, and can be manipulated multiple times. Therefore it is possible to generate as many alterations in a strain as desired. In addition, polar effects on genes downstream of the modification site can be minimized, allowing more precise modification of the organism13.
To generate mutant strains, suicide plasmids containing two DNA fragments identical to regions in the cyanobacterial chromosome flanking the gene to be deleted (termed the 5' and 3' flanking regions) are first constructed. Two genes are then inserted between these flanking regions. One of these encodes an antibiotic resistance protein; the second encodes SacB, which produces levansucrase, a compound conferring sensitivity to sucrose. In the first stage of the process, marked mutants, i.e. strains containing some foreign DNA, are generated. The plasmid construct is mixed with the cyanobacterial cells and the DNA is taken up naturally by the organism. Transformants are selected by growth on agar plates containing the appropriate antibiotic and the mutant genotype verified by PCR. Suicide plasmids cannot replicate within the strain of interest. Therefore any antibiotic resistant colonies will result from a recombination event whereby the gene of interest in inserted into the chromosome. To generate unmarked mutants, the marked mutant is then mixed with a second suicide plasmid containing just the 5' and 3' flanking regions. However, if insertion of foreign DNA is required, a plasmid consisting of the 5' and 3' flanking regions with a cassette containing the genes of interest inserted between these DNA fragments, can be used. Selection is via growth on agar plates containing sucrose. As sucrose is lethal to cells when the sacB gene product is expressed, the only cells that survive are those in which a second recombination event has occurred, whereby the sucrose sensitivity gene, in addition to the antibiotic resistance gene, has been recombined out of the chromosome and onto the plasmid. As a consequence of the recombinational exchange, the flanking regions and any DNA between them are inserted into the chromosome.
We have successfully used these methods to generate multiple chromosomal mutations in the same strain of Synechocystis sp. PCC6803 (hereafter referred to as Synechocystis)13,14, to introduce single point mutations into a gene of interest13 and for expression of gene cassettes. While generation of unmarked knockouts has been demonstrated prior to our work in Synechocystis15,16, a detailed method, aided by a visual presentation of the critical steps, is not publicly available. We have also applied the same method for generation of marked knockouts in another model cyanobacterium, Synechococcus sp. PCC7002 (hereafter referred to as Synechococcus). This protocol provides a clear, simple method for generating mutants and a rapid protocol for validating and storing these strains.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Preparation of Culture Media
- Prepare BG11 medium according to Castenholz, 198817.
- Prepare stock solutions of 100x BG11, trace elements and iron stock (Table 1).
- Prepare separate solutions of phosphate stock, Na2CO3 stock, N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (TES) buffer and NaHCO3 (Table 1).
- Autoclave the phosphate and Na2CO3 stocks. Filter-sterilize TES buffer and NaHCO3 with 0.2 µm filters.
- Prepare BG11 by combining 976 ml of water, 10 ml of 100x BG11, 1 ml of trace elements and 1 ml of iron stock and autoclave the solution. After this solution has cooled to room temperature, add 1 ml of phosphate stock, 1 ml of Na2CO3 stock and 10 ml of NaHCO3.
- For BG11 solid medium, add 15 g of agar and 700 ml of water to one flask. To the second flask, add 3 g of Na2S2O3, 226 ml of water, 10 ml of 100x BG11, 1 ml of trace elements and 1 ml of iron stock. Autoclave both solutions. After these solutions have cooled to room temperature, combine them and add 1 ml of phosphate stock, 1 ml of Na2CO3 stock, 10 ml of TES buffer, and 10 ml of NaHCO3.
Note: Solutions are prepared separately to avoid precipitation of certain salts.
- For selection on sucrose, prepare a 50% (w/v) sucrose solution. Filter sterilize the solution with 0.2 µm filters and add to BG11 (100 ml of 50% sucrose to 900 ml of BG11) to produce BG11/5% sucrose plates.
Note: Do not add NaHCO3 to BG11/5% sucrose agar plates. Add Na2CO3 as normal. - For culturing of Synechococcus add 10 ml of 1 M 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) and 1 ml of vitamin B12 (Table 1) to 1 L of BG11 medium.
Note: Transformation of strains cultured in commercially available BG11 media is significantly less efficient than in the BG11 media recipes described here and therefore is not recommended.
2. Growth of Cyanobacterial Strains
- Culture strains in 100 ml conical flasks with a maximum volume of 50 ml and shake at 120 rpm. Seal BG11 plates with Parafilm and puncture three small holes in the side of the plate to allow gas exchange. Incubate all strains at 30 °C under fluorescent bulbs in a photobioreactor at a light intensity between 20-40 µmol photons m-2 sec-1.
- Use best sterile techniques. Handle all cyanobacterial strains in a laminar flow hood.
Note: This is especially important when strains are cultured with media containing sucrose, which can be easily contaminated.
3. Generation of Plasmid Constructs
- Design sets of primers, including the required restriction enzyme sites, using primer design software such as Primer3 (http://frodo.wi.mit.edu/primer3/), to amplify two ~1 kb regions 5’ and 3’ of the gene of interest. Consult the genome sequence of the cyanobacterial species via Cyanobase (http://genome.kazusa.or.jp/cyanobase). See Table 2 for all primers used here. When designing primers consider the following factors:
- Ensure that amplified regions include 5' and 3' regions of the gene that will be mutated, e.g. Figure 1.
- Do not mutate intergenic regions to avoid unintended mutation of antisense and non-coding RNAs. For generation of mutants in Synechocystis, refer to the list of transcriptional start sites documented in Mitschke et al., 201118, in order to avoid mutation of antisense or non-coding RNAs.
- When choosing flanking regions do not include the entire open reading frame of adjacent genes as expression of these genes in Escherichia coli may interfere with cloning.
- Amplify products by PCR using high fidelity DNA polymerase according to the manufacturer's instructions.
Note: In our experience this enzyme produces few errors.- Set up 50 µl PCR reactions containing HF buffer and either 0, 1.5 or 3 µl of DMSO. Use 100 ng of genomic DNA per reaction. Use a program consisting of an initial denaturation step of 98 °C for 30 sec, 35 rounds of 98 °C for 10 sec, 67 °C for 30 sec, 72 °C for 30 sec, followed by a final extension step of 72 °C for 5 min. This typically gives consistent products.
- Verify PCR products and samples digested with endonuclease enzymes for the correct size via gel electrophoresis. Run 1% (w/v) agarose gels containing 0.02% (v/v) ethidium bromide for 45 min at 100 V.
CAUTION: Ethidium bromide is a potential mutagen and should be handled with appropriate protection. - Purify PCR products using a DNA purification kit according to the manufacturer's instructions. Also use this kit for purification of plasmid fragments, including pieces cut from agarose gels. Elute purified DNA in 14 µl of water.
- For cloning steps, incubate restriction endonuclease reaction mixtures at 37 °C for >1 hr in a total volume of 30 µl according to the manufacturer's instructions.
- For ligation steps, ligate DNA fragments at room temperature for >1 hr in a total volume of 20 µl, containing 5 µl of purified digested plasmid, 12 µl of purified digested insert, 2 µl of buffer and 1 µl of ligase.
- Prepare Escherichia coli DH5α transformant cells according to the following method.
- Grow an overnight E. coli culture in 10 ml Luria Bertani (LB) media.
- Inoculate 400 ml LB in a 1 L conical flask containing 6 ml 1 M MgCl2 (Table 1) with 1 ml of overnight culture.
- Grow the culture at 37 °C at 220 rpm for approximately 4 hr or until OD600nm reaches 0.4-0.6.
- Place cells on ice for 1 hr.
- Centrifuge at 2,800 x g for 10 min to pellet cells at 4 °C.
- Remove supernatant and resuspend in 160 ml solution A (Table 1) and incubate on ice for 20 min.
- Centrifuge at 2,800 x g for 10 min to pellet cells at 4 °C.
- Remove supernatant and resuspend in 4 ml Solution A + glycerol (Table 1).
- Prepare 50 µl aliquots, freeze in liquid N2, store at -80 °C.
- Mix 5 µl of ligation mixture with 50 µl of competent cells and incubate for 1 hr on ice.
- Heat shock the cells at 42 °C for 90 sec, followed by incubation on ice for 2 min.
- Add 950 µl of LB media (Table 1) and incubate at 37 °C for 1 hr.
- Aliquot 50 and 200 µl on plates with the appropriate antibiotic, either ampicillin (100 µg/ml) and/or kanamycin (30 µg/ml).
CAUTION: Both kanamycin and ampicillin are toxic and should be handled with appropriate protection. - Pick and incubate single colonies in 2 ml LB media inoculated with the appropriate antibiotic.
- Purify all plasmids using a miniprep plasmid purification kit according to the manufacturer's instructions.
- Generate plasmids, in this specific example for knocking out the cpcC1C2 genes, according to the following steps.
- Amplify the 1,012 bp 5' flanking region (left fragment) using primers cpcC1C2leftfor and cpcC1C2leftrev (See step 3.2, Table 2). Remove a small amount of the PCR reaction and confirm whether the correct size product has been amplified via gel electrophoresis (step 3.3). Digest this fragment and pUC19 with XbaI and BamHI (step 3.5).
- Purify both preparations (step 3.4), ligate (step 3.6), transform (step 3.7) and set up four 2 ml LB liquid cultures with ampicillin (100 µg/ml) from separate colonies for plasmid purification via minipreps (step 3.8).
- Check for insertion of the fragment into pUC19 via XbaI/BamHI digestion and gel electrophoresis (step 3.3). Bands of 2,660 bp and 1,012 bp indicate correct introduction of the insert into the plasmid.
- Amplify the 1,016 bp 3' flanking region (right fragment) using primers cpcC1C2rightfor and cpcC1C2rightrev (See step 3.2, Table 2). Remove a small amount of the PCR reaction and confirm whether the correct size product has been amplified via gel electrophoresis (step 3.3). Digest this fragment and pUC19 with SacI and EcoRI (step 3.5).
- Purify both preparations (step 3.4), ligate (step 3.6), transform (step 3.7) and set up four 2 ml LB liquid cultures with ampicillin (100 µg/ml) from separate colonies for plasmid purification via minipreps (step 3.8).
- Check for insertion of the fragment into pUC19 via SacI/EcoRI digestion (step 3.5) and gel electrophoresis (step 3.3). Bands of 2,660 bp and 1,016 bp indicate correct introduction of the insert into the plasmid.
Note: XbaI/BamHI sites for cloning of the 5' region and SacI/EcoRI for cloning of the 3' region into pUC19 are used wherever possible. If feasible, always include a BamHI site on the reverse primer for the 5' region or the forward primer for the 3' region to ensure that later cloning steps are easier to perform. - Sequence both inserts to determine if the sequence is correct using primers spanning the insertion site, e.g. M13 forward and M13 reverse (Table 2). The sequence must be correct to ensure no errors are introduced into flanking regions.
- Excise the left fragment from pUC19 via XbaI/BamHI digestion. Digest the pUC19 + right fragment with XbaI/BamHI (step 3.5).
- Purify the 1,012 bp left fragment and 3,676 bp pUC19 + right fragment from an agarose gel (step 3.3) via excision of the DNA using a scalpel blade.
- Purify both preparations (step 3.4), ligate (step 3.6), transform (step 3.7) and set up four 2 ml LB liquid cultures with ampicillin (100 µg/ml) from separate colonies for plasmid purification via minipreps (step 3.8).
- Check for insertion of the fragment into pUC19 + right fragment via XbaI/BamHI digestion (step 3.5) and gel electrophoresis (step 3.3). Bands of 3,676 bp and 1,012 bp indicate correct insertion of the insert into the plasmid (refer to this as plasmid B).
- Excise the npt1/sacB cassette from pUM24cm19 via BamHI digestion. Digest plasmid B with BamHI (step 3.5).
Note: The npt1/sacB cassette does not have to be purified from agarose gels since pUM24cm encodes a protein conferring chloramphenicol resistance. Therefore if colonies are grown on LB/ampicillin/kanamycin agar plates the only possible combination that will lead to resistant colonies is incorporation of the npt1/sacB cassette into plasmid B. - Purify both preparations (step 3.4), ligate (step 3.6), transform (step 3.7) and set up four 2 ml LB liquid cultures with ampicillin (100 µg/ml) and kanamycin (30 µg/ml) from separate colonies for plasmid purification via minipreps (step 3.8).
- Check for insertion of the npt1/sacB cassette into plasmid B via BamHI digestion (step 3.5) and gel electrophoresis (step 3.3). Bands of 4,688 bp and 3,894 bp indicate correct insertion of the insert into the plasmid (refer to this as plasmid A).
- Alternatively, blunt end the npt1/sacB cassette and clone into a different restriction endonuclease site between the left and right fragments in plasmid B. The npt1/sacB cassette must be cloned between the left and right fragments.
Note: If expression of a foreign cassette is required then this should be inserted between the left and right fragments of plasmid B. This plasmid is then used in the unmarked knockout steps.
4. Generation of Marked Synechocystis and Synechococcus Mutants
- Set up a fresh culture by inoculating a loop full of cells into 30-50 ml of BG11 medium. Grow the culture for 2-3 days to OD750nm = 0.2 to 0.6.
Note: Typically individual colonies are too small to use for inoculation and exposure of individual cells to even low levels of light will result in photoinhibition and selection for light resistant mutants. - Centrifuge 1-2 ml of the culture at 2,300 x g for 5 min and discard the supernatant. Do not centrifuge any cyanobacterial cultures at >2,300 x g as this may damage the cells. Wash the pellet once with BG11 medium.
Note: Do not resuspend cells by vortexing as this may result in loss of pili which are essential for DNA uptake. Resuspend cells by gentle pipetting. - Add BG11 medium to a final volume of 100 µl. Transfer cells to a 14 ml round-bottom tube.
- Add 1 µg of plasmid A to the cells and mix by gentle tapping. Add <10 µl of plasmid.
Note: Preferably the plasmid should be at a concentration of >100 ng/µl but concentrations lower than this are adequate for successful transformation. - Lay tubes down horizontally in the incubator. Incubate cultures for 4-6 hr.
Note: Cells can be briefly mixed by tapping every 1-2 hr but this is not essential. Samples can be placed in a shaking incubator although this does not significantly improve efficiency. - Spread aliquots of the cell culture/plasmid DNA mixture on BG11 agar plates without antibiotics. Typically 20 µl and 80 µl aliquots are spread on separate plates.
- ~24 hr later, add 2.5-3 ml of 0.6% Agar solution in water containing kanamycin (per 20 ml: 0.12 g of agar, 100 µl of 100 mg/ml kanamycin) to the agar plate. Cool this solution to ~42 °C, and add to the edge of the agar plate. Tilt the plate so the solution forms an even 'top agar' layer on the surface.
- Incubate agar plates for a further period of time. Colonies should be visible after approximately 7 days.
Note: Agar plates can be stacked 3 high in an incubator. Typically hundreds of colonies are obtained per transformation. - Streak individual colonies on BG11 + kanamycin (30 µg/ml) agar plates. Divide the agar plate into 6 sectors and use a blunt end toothpick to streak out the colonies over each individual sector. Obtaining single colonies is not important, just growth of the transformants.
- Confirm marked knockout by PCR using Taq DNA polymerase according to the manufacturer's instructions. Add 2 µl of MgCl2 (25 mM) per reaction.
- Remove a small proportion of the cells and transfer into a tube containing 50 µl water and ~20 425-600 µm glass beads. Shake in a vibrator for 5 min at ~2,000 rpm. Centrifuge at 15,700 x g for 5 min and use 5 µl of supernatant per 50 µl PCR reaction.
Note: Do not resuspend the solution. The cell debris needs to stay at the bottom of the tube.
- Remove a small proportion of the cells and transfer into a tube containing 50 µl water and ~20 425-600 µm glass beads. Shake in a vibrator for 5 min at ~2,000 rpm. Centrifuge at 15,700 x g for 5 min and use 5 µl of supernatant per 50 µl PCR reaction.
- Validate mutants
- Design primers which span the knockout region using primer design software (such as Primer3). Design primers starting at ~200 bp either side of the knockout region.
Note: Primers for verifying the cpcC1C2 mutant are outlined in Table 2 and are termed cpcC1C2for and cpcC1C2rev. - Amplify products using a program consisting of an initial denaturation step of 95 °C for 2 min, 35 rounds of 95 °C for 1 min, 60 °C for 1 min, 72 °C for 1 min per kb of sequence, followed by a final extension step of 72 °C for 5 min. Include a wild-type control. This typically gives consistent products.
- Verify the genotype via gel electrophoresis. Marked knockout transformants will show a band of ~4 kb (0.2 kb from both the left and right fragments plus the npt1/sacB cassette) and the absence of the wild-type band (Figure 2).
Note: In certain cases a ~4 kb band is not observed in the marked mutant due to the large size of this PCR product. However, if a band corresponding to the expected size of the wild-type is not observed then typically this strain is a marked knockout.
- Design primers which span the knockout region using primer design software (such as Primer3). Design primers starting at ~200 bp either side of the knockout region.
- If a wild-type band is still present then re-streak the strain on a fresh BG11 + kanamycin (30 µg/ml) agar plate and repeat the PCR. Repeat the re-streaking process until the mutant is segregated so that no wild-type band is observed in the PCR reaction.
Note: Increasing the amount of kanamycin to a concentration of 50 µg/ml, then 100 µg/ml is sometimes essential in order to segregate a marked mutant fully. - If the strain shows a marked mutant profile via PCR, then re-streak on a fresh BG11 + kanamycin (30 µg/ml) agar plate. Use this strain to generate the unmarked knockout.
Note: The protocol can be used to generate marked mutants with just an antibiotic resistance cassette. i.e. by replacing the npt1/sacB cassette with just the npt1 cassette from pUC18K20 between the left and right fragments.
5. Generation of Unmarked Synechocystis Mutants
- Set up a fresh culture of the marked knockout by inoculating a loop full of cells into 30-50 ml of BG11 medium. Grow the culture for 2-3 days to OD750nm = 0.2 to 0.6.
- Centrifuge 10 ml of the culture at 2,300 x g for 5 min and discard the supernatant. Wash once with BG11 medium.
Note: Do not resuspend cells by vortexing as this may result in loss of pili which are essential for DNA uptake. Resuspend cells by gentle pipetting. - Add BG11 to a final volume of 200 µl. Transfer cells to a 14 ml round-bottom tube.
- Add 1 µg of plasmid B DNA to the cells and mix by gentle tapping.
- Incubate the samples for 4-6 hr. Lay tubes down horizontally.
Note: Cells can be briefly mixed by tapping every 1-2 hr but this is not essential. Samples can be placed in a shaking incubator although this does not improve efficiency. - Add 1.8 ml of BG11 medium and incubate samples for a total of 4 days with shaking. This is sufficient time to allow recombination to occur in the multiple chromosomal copies.
- Plate aliquots of the transformation mixture on BG11/5% sucrose agar plates. Plate 50 µl, 10 µl and 1 µl per agar plate. If a colony lawn appears on all these agar plates dilute the solution further and aliquot on fresh plates. Colonies should be visible after approximately 7 days.
- Patch 30-50 individual colonies on BG11 + kanamycin (30 µg/ml) agar plates first and BG11/5% sucrose agar plates second, using a blunt end toothpick. Any bacteria that grow on BG11/5% sucrose plates but not BG11 + kanamycin plates are potential unmarked knockouts. Bacteria growing on both plates are likely to be sucrose resistant due to a mutation in the sacB gene.
- Verify unmarked knockouts using the same primers and method as was used to check the marked knockouts. e.g. cpcC1C2for and cpcC1C2rev (Table 2) for verifying the cpcC1C2 unmarked knockout. An unmarked knockout will show a band on an agarose gel corresponding to the wild-type size minus the deleted region (Figure 2).
- If the strain shows an unmarked mutant profile via PCR (step 4.11.2) and gel electrophoresis (Figure 2), then re-streak on a fresh BG11 agar plate without antibiotics.
6. Long-term Storage of Strains
- Set up a fresh culture of the strain by inoculating a loop full of cells into 30-50 ml of BG11 medium. Grow the culture for 3-4 days to OD750nm = 0.4 to 0.7.
- Wash cells once with BG11 and resuspend in ~2 ml of BG11.
- Add 0.8 ml of concentrated cells to one tube. Then add 0.2 ml of 80% filter sterilized glycerol.
- Optional: Add 0.93 ml of concentrated cells to another tube. Add 0.07 ml of DMSO to this tube.
CAUTION: DMSO is toxic and should be handled with appropriate protection. - Store both tubes at -80 °C. To revive strains remove the tube and scrape off some cells with a blunt toothpick onto an agar plate without antibiotics. Streak out as normal using a sterile loop.

Figure 1: Plasmid construction for generation of marked and unmarked knockouts, e.g. cpcC1 and cpcC2 in Synechocystis. (A) Region of the Synechocystis genome where (B) cpcC1 and cpcC2 and adjacent genes are located. Highlighted in black is the region of the genome to be deleted in the mutant. (C) Sites of the genome which are amplified by PCR. The 5' flanking region (indicated in blue) and 3' flanking region (indicated in red) are amplified with restriction endonuclease sites for cloning into pUC19. The 5' (or 3') flanking region is excised out of pUC19 and inserted into the pUC19 + 3' (or 5') flanking region plasmid to generate plasmid B. (D) The npt1/sacB cassette from pUM24 is excised via BamHI digestion and inserted between the 5' and 3' flanking regions to generate Plasmid A. Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Plasmid design is critical for successful generation of both marked and unmarked mutants. Figure 1 gives an example of plasmid A and B used to generate a deletion mutant in the Synechocystis genes cpcC1 and cpcC213. In each case the 5' and 3' flanking regions are approximately 900-1,000 bp. Reduced flanking regions can be used although the smallest we have successfully trialed has been approximately 500 bp. Plasmid B can also contain a gene cassette between the 5' and 3' ~1 kb flanking regions or a modified version of the native gene sequence.
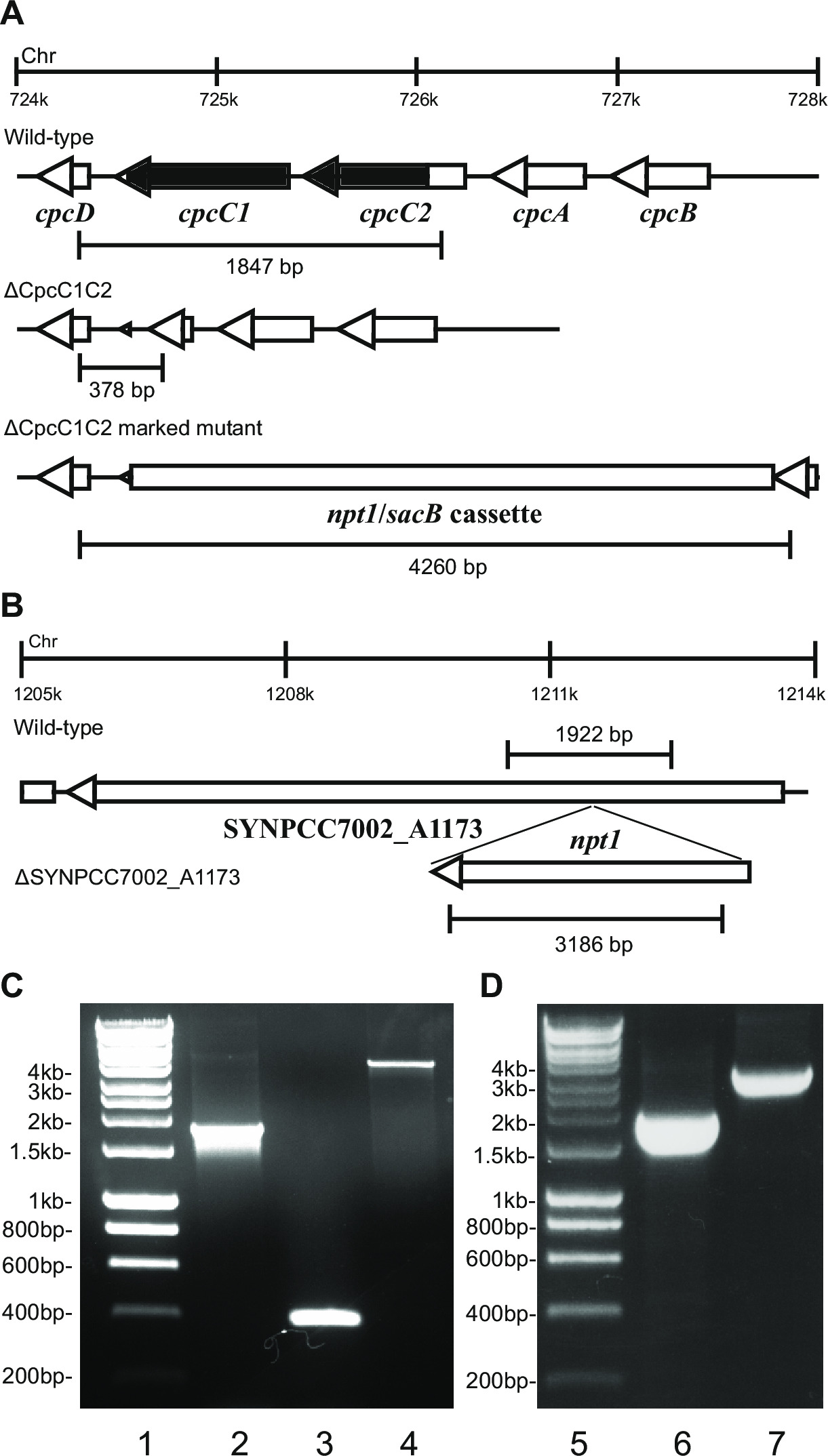
Figure 2: Verification of marked and unmarked mutants, e.g. cpcC1/cpcC2 in Synechocystis and SYNPCC7002_A1173 in Synechococcus. (A) The expected size of the wild-type Synechocystis (top), unmarked (middle) and marked knockout (bottom) amplicons generated using primers cpcC1C2for and cpcC1C2rev, approximately 200 bp on either side of the chromosomal region to be deleted. (B) The expected size of the wild-type Synechococcus (top) and marked knockout (bottom) amplicons generated using primers A1173for and A1173rev, approximately 200 bp on either side of the chromosomal region to be deleted. Agarose gel showing amplicons generated from (C) wild-type Synechocystis (lane 2), unmarked (lane 3) and marked cpcC1/cpcC2 knockouts (lane 4), and (D) wild-type Synechococcus (lane 6) and the marked SYNPCC7002_A1173 knockout (lane 7). Markers are shown in lanes 1 and 5. Please click here to view a larger version of this figure.
Upon transformation of plasmid A into the cells, typically several hundred colonies will appear on a plate after approximately 7-10 days. Colonies are <1 mm in diameter and will not increase in size for the next few weeks. Therefore it is critical to use a blunt end toothpick to remove the colony and streak it on a fresh BG11 + kanamycin agar plate. Approximately half the re-streaked colonies will grow after 4-6 days. If genes are non-essential and mutants demonstrate growth similar to the wild-type strain under continuous light of 20-40 µmol photons m-2 sec-1 (e.g. terminal oxidase mutants in Lea-Smith et al., 201314) (Figure 3), then all the chromosomes should contain a copy of the npt1/sacB cassette inserted sequence, as determined via PCR. If genes are non-essential and mutants demonstrate a slow growth phenotype under continuous light of 20-40 µmol photons m-2 sec-1 (e.g. phycobilisome deficient mutants in Lea-Smith et al., 201413) (Figure 3), then several rounds of re-streaking on BG11 agar plates with gradually increased amounts of kanamycin are essential in order to obtain a segregated marked mutant. Once a segregated mutant is obtained this should be re-streaked on a fresh BG11 plus kanamycin agar plate to ensure that segregation is complete. If repeated rounds of streaking do not result in a segregated marked mutant then the gene is likely essential for survival. Figure 4 gives an outline of the experimental steps involved in unmarked mutant generation.
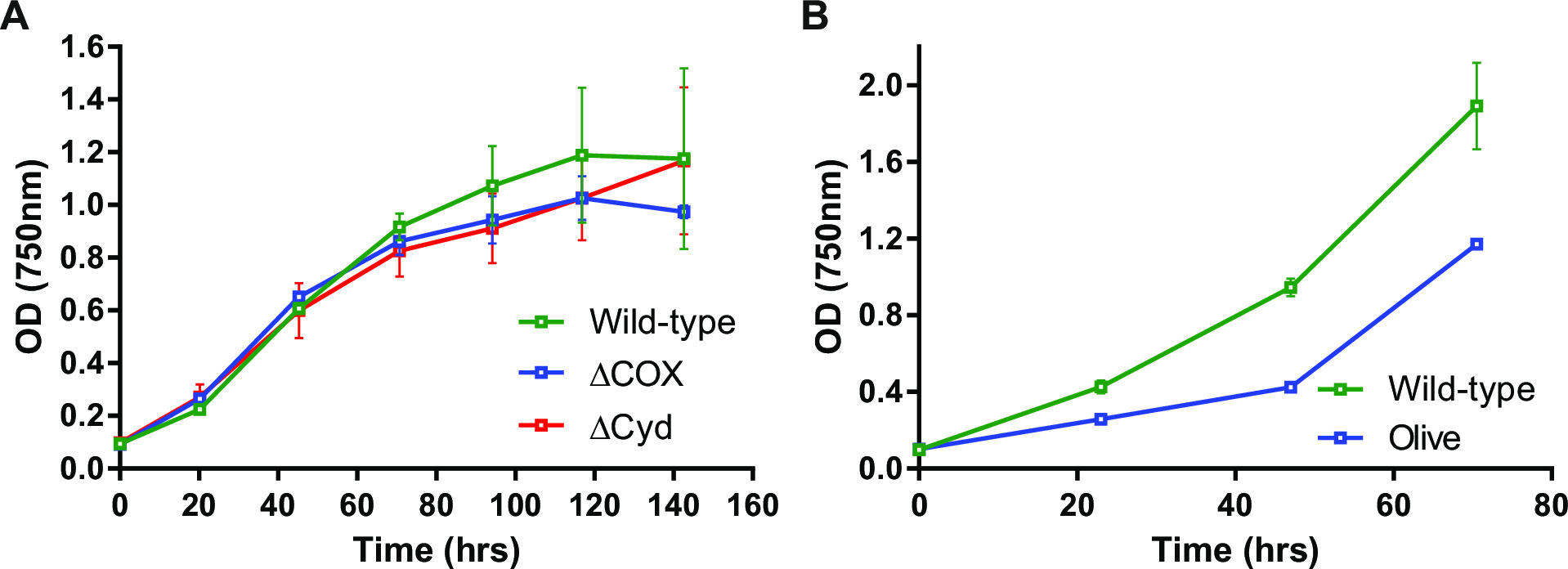
Figure 3: Growth of Synechocystis mutants. Examples of mutants which demonstrate (A) similar growth to wild-type and (B) slower growth than wild-type. The ΔCOX mutant lacks cytochrome oxidase due to deletion of the CtaC1D1E1 genes. The ΔCyd mutant lacks quinol oxidase due to deletion of the CydAB genes. The olive mutant lacks a portion of the phycobilisome due to deletion of the CpcABC1C2D genes. Samples in (B) were bubbled with air to facilitate growth. Reproduced from data published in Lea-Smith et al., 201314 and 201413 (www.plantphysiol.org; Copyright American Society of Plant Biologists). Please click here to view a larger version of this figure.
Generation of unmarked mutants is highly efficient. Upon transformation of plasmid B into the marked mutant, a four day incubation period and subsequent plating on BG11 plus sucrose agar plates, hundreds of colonies are obtained per 1-10 µl of transformed cell suspension. However a series of dilutions should be trialed, ranging from 0.1 to 100 µl, as an excessive amount leads to a reduced concentration of sucrose per cell, resulting in poor selection for unmarked mutants. If a lawn of cells is seen over the entire plate then lower concentrations should be tried. Once individual colonies are obtained on plates, patching on BG11 plus sucrose and BG11 plus kanamycin agar plates is an essential step. Typically for unmarked mutants where a region of the chromosome is being deleted, the majority of colonies will be kanamycin sensitive and sucrose resistant. PCR amplification of the target region in these colonies show that nearly 100% demonstrate the unmarked mutant profile, e.g. Figure 2. If a gene cassette is being inserted into the chromosome then typically a higher proportion of kanamycin resistant and sucrose resistant colonies are observed. These mutants can grow on sucrose due to a mutation in the sacB gene. If no kanamycin sensitive and sucrose resistant colonies are generated then the gene cassette is deleterious to the cell.

Figure 4: Generation of marked and unmarked mutants in Synechocystis. Schematic detailing (A) recombination and (B) experimental steps involved in mutant generation. Plasmid A is first mixed with cells. Following incubation on agar plates containing kanamycin, colonies in which a recombination event occurs between the 5' and 3' flanking regions (indicated in blue and red, respectively) and the homologous sequence in the chromosome, are isolated. In addition, the npt1/sacB cassette between the 5' and 3' flanking regions is inserted into the chromosome. Following segregation a marked mutant is generated. Marked mutant cells are then mixed with plasmid B which can contain either (C) 1: the 5' and 3' flanking regions; 2: the 5' and 3' flanking regions with an expression cassette containing genes of interest inserted between these sequences; 3: the 5' and 3' flanking regions with the wild-type sequence with the desired nucleotide alterations inserted between these sequences. A second homologous recombination event occurs between the 5' and 3' flanking regions and the homologous regions in the chromosome, resulting in removal of the npt1/sacB cassette and either the unmarked knockout or a mutant with an insertion or altered wild-type region introduced into the chromosome. Please click here to view a larger version of this figure.
| Stock solution recipes | |
| Chemical | Amount (g) |
| 100x BG11 (per L) | |
| NaNO3 | 149.6 |
| MgSO4.7H2O | 7.49 |
| CaCl2.2H2O | 3.6 |
| Citric acid | 0.6 |
| Add 1.12 ml 0.25 M Na2EDTA, pH 8.0 | |
| 0.25 M Na2EDTA, pH 8.0 (per 100 ml) | |
| Na2EDTA | 9.3 |
| Trace elements (per 100 ml) | |
| H3BO3 | 0.286 |
| MnCl2.4H2O | 0.181 |
| ZnSO4.7H2O | 0.022 |
| Na2MoO4.2H2O | 0.039 |
| CuSO4.5H2O | 0.008 |
| Co(NO3)2.6H2O | 0.005 |
| Iron stock (per 100 ml) | |
| Ferric ammonium citrate | 1.11 |
| Phosphate stock (per 100 ml) | |
| K2HPO4 | 3.05 |
| Na2CO3 stock (per 100 ml) | |
| Na2CO3 | 2 |
| TES buffer, pH 8.2 (per 100 ml) | |
| TES | 22.9 |
| NaHCO3 stock (per 100 ml) | |
| NaHCO3 | 8.4 |
| HEPES, pH 8.2 (per 500 ml) | |
| HEPES | 119.15 |
| Vitamin B12 (Per 50 ml) | |
| Cyanocobalamin | 0.02 |
| Luria Bertani media (Per 500 ml) | |
| Luria Bertani broth | 12.5 |
| 1 M MgCl2 (Per 100 ml) | |
| MgCl2.6H2O | 20.33 |
| Solution A (Per 200 ml) | |
| MnCl2.4H2O | 0.395 |
| CaCl2.2H2O | 1.47 |
| 2-(N-Morpholino)ethanesulfonic acid hydrate, 4-Morpholineethanesulfonic acid (MES) | 0.4265 |
| Solution A + glycerol | |
| 10 ml solution A | |
| 1.5 ml glycerol |
Table 1: Solutions used in this study.
| Primer | Sequence |
| cpcC1C2leftfor | GTACTCTAGAGCGGCTAAATGCTACGAC |
| cpcC1C2leftrev | GATCGGATCCGCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATCGAGCTCTGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACTGAATTCATCGTTGCTTGAACGGTCTC |
| M13 forward | TGTAAAACGACGGCCAGT |
| M13 reverse | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Table 2: Primers used in this study. Restriction endonuclease sites are underlined.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
The most critical steps in generation of unmarked mutants are: 1) careful plasmid design to ensure only the targeted region is altered; 2) ensuring that samples remain axenic, especially when cultured on sucrose; 3) plating transformed cells for marked mutant generation initially on BG11 agar plates lacking antibiotics, followed by addition of agar plus antibiotics 24 hr later; 4) culturing marked mutants for 4 full days prior to plating on BG11 plus sucrose agar plates: 5) ensuring that marked mutants are fully segregated and 6) thoroughly confirming the genotype of mutant strains. For this last step, additional primers designed to amplify part of the deleted region, can be used to ensure that it has been removed. Southern blotting, while laborious, can also be used. However, our experience is that the procedure outlined in this paper is sufficient for proper verification of mutants. This procedure has also been used to generate marked mutants in Synechococcus elongatus PCC7942. However, repeated transformation of this cyanobacterium has proved challenging.
If marked mutants cannot be segregated then different environmental conditions high CO2, low light (<20 µmol photons m-2 sec-1) or additional nutrients (i.e. glucose) can be tested. For example, the addition of glucose is essential in order to generate photosystem II mutants21. If marked mutants never fully segregate then the gene is probably essential for viability. However, there are examples from the literature where some research groups have been unable to knockout a gene (For example, Vipp in Synechocystis)22, only for other groups to later show that the gene is not essential23. This could be due to differences in the wild-type strains or incorrect plasmid design, resulting in polar effects on adjacent, essential genes. If a mutant does not fully segregate we would recommend that the plasmid containing the npt1 cassette from pUC18K20 between the left and right fragments be used for transformation. It is easier to verify the presence of bands corresponding to the wild-type and mutant by PCR, since this fragment is approximately 1.2 kb, compared to the 3.8 kb npt1/sacB cassette. This result is an important piece of evidence demonstrating that the gene is essential.
Generation of unmarked mutants with inserted expression cassettes is generally more challenging than development of knockout strains. We generally express genes under control of the strong cpcBAC1C2D promoter13. In some cases this may decrease the chances of successful insertion of the gene cassette, if over-expression of a protein is deleterious to the cell. Weaker promoters should then be tested. In general we have observed that the larger the gene cassette is, the more difficult it is to insert it into the genome. We have not been able to insert gene cassettes larger than 5 kb. Care must also be taken in choosing sites to insert expression cassettes into the genome. Neutral sites that do not affect cell viability or growth should be used. Examples in Synechocystis include phaAB and phaCE, which encode the proteins encoding the polyhydroxybutyrate biosynthetic pathway24,25. More recently an extensive list of neutral sites in Synechocystis has been identified26.
Generation of unmarked mutants in cyanobacteria is a slow process, taking approximately 5-7 weeks if all steps are conducting properly. This is slower than the standard method of generating marked knockouts utilized by the majority of research groups investigating cyanobacteria. However, the flexibility of being able to introduce further mutations into unmarked mutants partially compensates for this, since additional plasmids containing a range of cassettes conferring resistance to different antibiotics, do not have to be constructed. For research purposes the ability to mutate multiple genes is sometimes necessary in order to fully characterize the functional role of proteins. For example, we identified a deleterious phenotype only upon deletion of the two terminal oxidase electron sinks localized to the thylakoid membrane, since loss of only one of these complexes could be compensated for by activity of the other14. Development of a strain for industrial applications will also require multiple modifications to a strain, not just for introduction of foreign genes but also to increase photosynthetic efficiency, light harvesting optimization and deletion of competing pathways for the desired substrate.
The major factor limiting the speed of unmarked mutant generation is the slow division time of model cyanobacterial species, between 8-20 hr depending on light conditions. Under higher light intensities and CO2 concentrations, growth is faster. However, there is a risk that mutant strains which cannot tolerate either high light or CO2 will be selected against, or that mutant strains will undergo undesirable alterations prior to phenotypic characterization. Therefore this is not recommended. However, it would be highly advantageous if a more rapid protocol to generate unmarked mutants was developed. Overall, this would facilitate the development of strains for both basic research and applied applications. Such strains could be used for biofuel, biomass or chemical production or in understanding many aspects of cyanobacterial biochemistry, genetics and physiology.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We are grateful to the Environmental Services Association Education Trust, the Synthetic Biology in Cambridge SynBio fund and the Ministry of Social Justice and Empowerment, Government of India, for financial support.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
