We developed an ASO-based RNA isolation strategy, termed TRIP, to capture particular mRNAs with their bound proteins from three different organisms37. Essentially, RNA-protein complexes were crosslinked in vivo by UV-irradiation of cells at 254 nm, and poly(A) RNAs were recovered with commercially available oligo (dT) coupled-magnetic beads, then the mRNA of interest was isolated with 3'-biotinylated 2-0'-methoxy modified antisense RNA oligonucleotides (Figure 1). We therefore designed several 21-24 nts modified ASOs with full-complementarity to regions in the selected mRNAs from yeast, C. elegans and human, and tested their suitability to recover the mRNA of interest (a list of primers and ASOs is given in Table 1). The efficiency and specificity of individual ASOs were first evaluated with non-crosslinked total RNA isolated from the respective organism. In these experiments, RNA ASOs were coupled to streptavidin-conjugated paramagnetic beads and incubated with non-crosslinked total RNA prepared from the corresponding organism. After the release of captured mRNAs from the beads, the presence of mRNA targets as well as unrelated control mRNAs was monitored by reverse transcription (RT)-polymerase chain reaction (PCR)37. We noticed that two variables, the salt concentration and temperature of wash buffers, played critical roles in the efficiency of the capture of PFK2 mRNA from yeast that was tested with three different ASOs (Figure 2A). Lowering the salt (NaCl) concentration to 25 mM reduced the recovery of the negative control mRNA (PFK1) to non-detectable levels with all ASOs, but it also reduced the recovery of the desired target mRNA PFK2 (10-15% of the input). Conversely, an increase of the salt concentrations to physiological levels (150 mM NaCl) increased the recovery of PFK2 mRNAs up to 75% with the ASO PFK2-2, exceeding that of the control PFK1 mRNA by at least 5-fold (Figure 2A). Of further note, the different ASOs showed great variation of mRNA target capture efficacies at physiological salt-concentrations, emphasizing the need for empirical validation of ASOs. The dependence of mRNA recovery on the temperature of the wash buffer is exemplified for C. elegans cep-1 and yeast RPS20 mRNAs, using respective ASOs (Table 1). We observed that the optimal wash temperature was between 50 °C and 55 °C, as evident from the low background with unrelated mRNAs and the efficient recovery of mRNAs target (Figure 2B). At this point,we wish to emphasize the possibility of cross-hybridization of ASOs with other mRNAs. For example, the DNM1 ASO is fully complementary to a sequence within the DNM1 coding sequence, but it also partially anneals with ACT1 mRNA. DNM1 ASOs recovered both mRNAs irrespective of the washing temperature, showing strong propensity for cross-hybridization (Figure 2C). Finally, we have used the above described tests and optimizations to select three ASOs that were suitable for the recovery of respective target mRNAs from total RNA isolated from S. cerevisiae, C. elegans and human cells (Figure 2D).
After initial selection of suitable ASOs in vitro, we performed TRIP with cell extracts obtained from UV-crosslinked organisms/cells (Figure 1). Specifically, we tested the recovery of three different mRNAs from three different organisms: PFK2 mRNA from the yeast S. cerevisiae, cep-1 from the nematode C. elegans, and a reporter mRNA (pGL3-CDKN1B-3'UTR) bearing the 3'UTR sequence of human CDKN1B/p27 mRNA fused to a luciferase (luc) reporter for transient expression in human HEK293 cells. To control the specificity of the RNA isolation, we monitored the recovery of several unrelated mRNAs, and we performed competition experiments by the addition of an excess of pA to cell extracts, which competes with the binding of cellular mRNAs to oligo(dT)25 beads during the first step purification step. As previously seen with non-crosslinked total RNA samples (Figure 2), RT-PCR confirmed the enrichment of the respective mRNAs target mRNA during TRIP, whereas several non-related control mRNAs were not enriched (Figure 3A). Moreover, neither mRNAs were detected on beads without ASOs, indicating appropriate blocking procedures that avoid unspecific binding. On this line, unrelated/scrambled ASOs may also be used as control, although the potential for cross-hybridization with certain mRNAs and the inferred capture of bound proteins has then to be taken into account. Since proteins in the final eluate could be hardly visualized on silver-stained polyacrylamide gels (data not shown), the presence of previously known mRNA interacting proteins was further assessed by immunoblot analysis. This includes Pfk2p from S. cerevisiae, which binds selectively to PFK2 mRNA in a ribosome independent manner12; C. elegans GLD-1, a canonical RBP that binds to 3'UTR sequences of cep-1 mRNA for translational regulation43; and HuR, an RBP that regulates mRNA stability and translation of p27/CDKN1B mRNA44. As expected, all of these proteins were identified in the TRIP eluates of respective mRNAs by Western Blot analysis (Figure 3B).
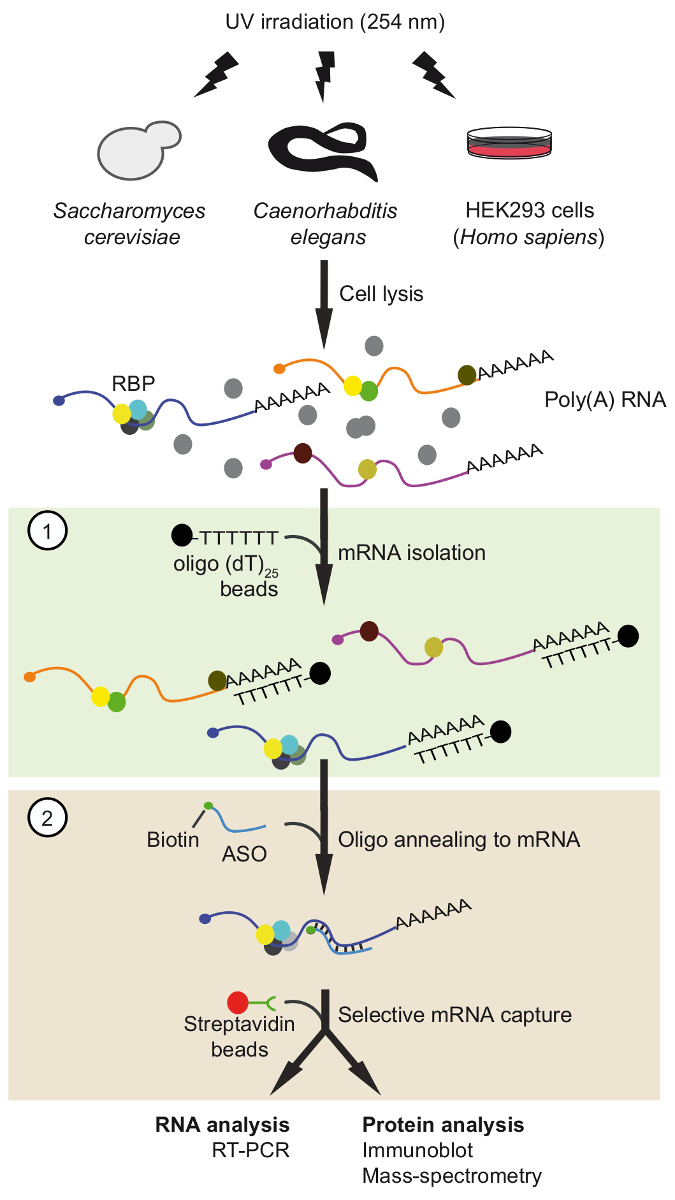
Figure 1. Schematic representation of TRIP. Proteins are crosslinked to RNA in vivo by UV-irradiation. In the first step (light green box), poly(A) RNA-protein complexes are recovered with oligo(dT)25 beads applying stringent washing conditions to remove unbound proteins. In the second step (pink box), the target mRNP is pulled-out with biotinylated antisense RNA oligonucleotides and streptavidin beads. The purified mRNPs are then analyzed by RT-PCR and immunoblot/mass-spectrometry (MS) to identify RNAs and proteins interacting with the mRNA of interest, respectively. The figure was modified from previous publication37 with permission. Please click here to view a larger version of this figure.

Figure 2. Isolation of selected mRNAs from total RNA of non-crosslinked cells/organisms using modified antisense RNA capture probes. (A) Impact of salt concentrations on the capturing efficiency of the yeast PFK2 mRNA with ASOs. Total RNA from yeast cells was combined with the indicated ASOs and washed with buffer containing the specified salt (NaCl) concentration at 55 °C. Green, blue and orange lines represent PFK2 mRNA recovery with different ASOs as determined by RT-PCR37: PFK2-1 and PFK2-2 anneal in the 3'UTR, PFK2-4 in the CDS. PFK1 is a negative control mRNA. PCR was performed with 32 amplification cycles and quantified as previously described37. (B) Fraction of C. eleganscep-1 and yeast RPS20 ASO bound mRNAs at different wash temperatures, represented in black and red lines, respectively. C. eleganspgk-1 and yeast ACT1 are non-target (control) mRNAs. 30 and 32 PCR cycles were applied for detection of yeast and C. elegans mRNAs, respectively. (C) Schematic representation of the hybridization of DNM1 ASO (red) with sequences in the DNM1 mRNA as well as potential cross-hybridization with ACT1 mRNA is shown to the left. An agarose gel showing products from RT-PCR reactions (30 cycles) for the detection of yeast DNM1 and ACT1 mRNAs eluted from beads is shown to the right. Input, total RNA; Sn, supernatant after incubation with ASOs; E, eluates from beads washed at indicated temperatures prior elution (40 °C, 50 °C and 60 °C). A control experiment (Ctrl) was performed in parallel without adding ASO. (D) Agarose gel showing RT-PCR products for the detection of mRNAs (right) captured from total RNA of yeast S. cerevisiae, C. elegans, and human HEK293 cells (H. sapiens). Input, total RNA; Sn, supernatant; E, eluates from the beads. PCR was performed by 30 amplification cycles for yeast mRNAs, 32 cycles for C. elegans mRNAs, and 28 and 30 cycles for human tubulin and p27 mRNAs, respectively. The figure was modified from previous publication37 with permission. Please click here to view a larger version of this figure.

Figure 3. Capture of specific mRNA-protein complexes from extracts derived from UV crosslinked cells with TRIP. (A) Agarose gels for the detection of mRNAs (right) with RT-PCR upon capture with oligo(dT) and indicated ASOs from S. cerevisiae, C. elegans and human (H. sapiens) cell extracts. Input, total RNA from crosslinked cells/organisms; Ctrl, control without ASO. Poly(A), competition with poly(A). RT-PCR was performed as described37 with 35 amplification cycles for LUC, 32 cycles for p27, and 29 cycles for tubulin. (B) Immunoblot analysis of mRNA-bound proteins with indicated antibodies (right). Loaded fractions are as follows: 0.1%, 2.5% and 1% for yeast, nematode and human inputs; 10%, 10% and 5% for yeast, nematode and human oligo(dT) lanes; and 66% for all ASOs lanes. Molecular weights (MW) are indicated in kilodaltons (kDa). Figure republished with permission37. Please click here to view a larger version of this figure.
| Primer name | Sequence | Target | Size | |
| Pfk2_Fwd | GTGTTAAGGGTTCACATGTCG | PFK2 S. cerevisiae | 133 bp | |
| Pfk2_Rev | CTTCCAACCAAATGGTCAGC | PFK2 S. cerevisiae | 133 bp | |
| Pfk1_Fwd | GGTGATTCTCCAGGTATGAATG | PFK1 S. cerevisiae | 97 bp | |
| Pfk1_Rev | CTTCGTAACCTTCGTAAACAGC | PFK1 S. cerevisiae | 97 bp | |
| Act1_Fwd | GTCTGGATTGGTGGTTCTATC | ACT1 S. cerevisiae | 85 bp | |
| Act1_Rev | GGACCACTTTCGTCGTATTC | ACT1 S. cerevisiae | 85 bp | |
| Dnm1_Fwd | CTGTGTTCGATGCATCAGAC | DNM1 S. cerevisiae | 156 bp | |
| Dnm1_Rev | CGCACTCCAATTCTTCTCTC | DNM1 S. cerevisiae | 156 bp | |
| Rps20_Fwd | CGCTGAACAACACAACTTGG | RPS20 S. cerevisiae | 228 bp | |
| Rps20_Rev | GGAAGCAACAACAACTTCGAC | RPS20 S. cerevisiae | 228 bp | |
| Cep1_Fwd | CGATGAAGAGAAGTCGCTGT | cep-1 C. elegans | 110 bp | |
| Cep1_Rev | ATCTGGGAACTTTTGCTTCG | cep-1 C. elegans | 110 bp | |
| Pgk1_Fwd | GCGATATTTATGTCAATGATGCTTTC | pgk-1 C. elegans | 74 bp | |
| Pgk1_Rev | TGAGTGCTCGACTCCAACCA | pgk-1 C. elegans | 74 bp | |
| Mpk1_Fwd | TGCTCAGTAATCGGCCATTG | mpk-1 C. elegans | 74 bp | |
| Mpk1_Rev | TCCAACAACTGCCAAAATCAAA | mpk-1 C. elegans | 74 bp | |
| p27_Fwd | TTTAAAAATACATATCGCTGACTTCATGG | p27 H. sapiens | 212 bp | |
| p27_Rev | CAAAGTTTATGTGCTACATAAAAGGTAAAAA | p27 H. sapiens | 212 bp | |
| Luc_Fwd | AATGGCTCATATCGCTCCTGGAT | Luciferase P. pγralis | 117 bp | |
| Luc_Rev | TGGACGATGGCCTTGATCTTGTCT | Luciferase P. pγralis | 117 bp | |
| β-TUBULIN_Fwd | CTGAACCACCTTGTCTCAGC | β-TUBULIN H. sapiens | 136 bp | |
| β-TUBULIN_Rev | AGCCAGGCATAAAGAAATGG | β-TUBULIN H. sapiens | 136 bp | |
| PFK2-1 ASO | AAUAGAAAGUGUAAUAAAAGGUCAU | 3' UTR PFK2 S. cerevisiae | – | |
| PFK2-2 ASO | GUUUCAUGGGGUAGUACUUGU | 3' UTR PFK2 S. cerevisiae | – | |
| PFK2-4 ASO | CUUGAAGAGGAGCGUUCAUA | ORF PFK2 S. cerevisiae | – | |
| DNM1 ASO | UCGGUCAGUGGAGGUUCAGCGUUU | ORF DNM1 S. cerevisiae | – | |
| RPS20 ASO | GUCGGUAAUAGCCUUCUCAUUCUUG | ORF RPS20 S. cerevisiae | – | |
| cep-1 ASO | GUGAGAAAUGCGGUGCUUUGAAA | 3' UTR cep-1 C. elegans | – | |
| p27 ASO | UCAUACCCCGCUCCACGUCAGUU | 3' UTR p27 H. sapiens | – | |
Table 1. Oligonucleotide sequences. List of PCR primers and ASOs used in this work, primer sequence, gene target and expected fragment size after amplification.
