

Summary
Cet article vise à fournir la méthodologie pour la transgenèse lentivirale chez les embryons de rats à l’aide d’injections multiples d’une suspension de virus dans l’espace périvitelline zygote. Les rats femelles qui sont accouplés avec une souche masculine fertile avec une couleur de fourrure dominante différente est employée pour générer des mères adoptives pseudo-enceintes.
Abstract
Les modèles animaux transgéniques sont fondamentalement importants pour la recherche biomédicale moderne. L’incorporation de gènes étrangers dans les premiers embryons de souris ou de rats est un outil inestimable pour l’analyse de la fonction génique dans les organismes vivants. La méthode standard de transgenèse est basée sur le microinjectage de fragments d’ADN étrangers dans un pronucléus d’un ocrayte fécondé. Cette technique est largement utilisée chez la souris, mais reste relativement inefficace et techniquement exigeante chez d’autres espèces animales. Le transgène peut également être introduit dans des embryons à un étage par infection lentivirale, offrant une alternative efficace aux injections pronucléaires standard, en particulier chez les espèces ou les souches ayant une structure embryonnaire plus difficile. Dans cette approche, une suspension qui contient des vecteurs lentiviraux est injectée dans l’espace périvitelline d’un embryon de rat fécondé, qui est techniquement moins exigeant et a un taux de réussite plus élevé. Il a été démontré que les vecteurs lentiviraux intègrent efficacement le transgène dans le génome pour déterminer la génération de lignées transgéniques stables. Malgré certaines limitations (p. ex., les exigences de biosécurité de niveau 2, les limites de taille des fragments d’ADN), la transgenèse lentiviral est une méthode de transgenèse rapide et efficace. En outre, l’utilisation de rats femelles qui sont accouplés avec une souche masculine fertile avec une couleur de fourrure dominante différente est présentée comme une alternative pour générer des mères adoptives pseudo-enceintes.
Introduction
Depuis de nombreuses années, les rongeurs de laboratoire, comme les rats et les souris, ont été utilisés pour modéliser les conditions physiologiques et pathologiques humaines. La recherche sur les animaux a mené à des découvertes qui étaient inaccessibles par tous les autres moyens. Initialement, les études génétiques se sont concentrées sur l’analyse des désordres spontanément et phénotypes qui sont considérés pour imiter étroitement la condition humaine1. Le développement de méthodes de génie génétique a permis l’introduction ou la suppression de gènes spécifiques pour obtenir un phénotype désiré. Par conséquent, la génération d’animaux transgéniques est reconnue comme une technique fondamentale dans la recherche moderne qui permet des études de la fonction génique dans les organismes vivants.
La technologie animale transgénique est devenue possible grâce à une combinaison de réalisations en embryologie expérimentale et en biologie moléculaire. Dans les années 1960, l’embryologiste polonais A. K. Tarkowski a publié les premiers travaux sur la manipulation des embryons de souris au cours des premiers stades du développement2. En outre, les biologistes moléculaires ont développé des techniques pour générer des vecteurs d’ADN (c.-à-d., porteurs) pour l’introduction, entre autres, de l’ADN étranger dans le génome de l’animal. Ces vecteurs permettent la propagation de gènes sélectionnés et leur modification appropriée, selon le type de recherche qui est menée. Le terme « animal transgénique » a été introduit par Gordon et Ruddle3.
La première espèce largement acceptée qui a été utilisée dans la neurobiologie, la physiologie, la pharmacologie, la toxicologie, et beaucoup d’autres domaines des sciences biologiques et médicales était le rat de Norvège, Rattus norvegicus4. Cependant, en raison de la difficulté à manipuler les embryons de rats, la souris maison Mus musculus est devenue l’espèce animale dominante dans la recherche génétique5. Une autre raison de la primauté de la souris dans une telle recherche était la disponibilité de la technologie des cellules souches embryonnaires pour générer des animaux knock-out pour cette espèce. La technique la plus couramment utilisée de transgenèse (2 à 10 % de la progéniture transgénique par rapport à tous les animaux nés) est la microinjection de fragments d’ADN dans un pronucléus d’un oocyte fécondé. En 1990, cette approche, qui a été introduite pour la première fois chez la souris, a été adaptée pour les rats6,7. La transgenèse de rat par injection pronucléaire est caractérisée par une efficacité inférieure8 comparée aux souris, qui est strictement liée à la présence de plasma élastique et de membranes pronucléaires9. Bien que la survie des embryons après manipulation soit de 40 à 50 % inférieure à celle des souris, cette technique est considérée comme une norme dans la génération de rats génétiquement modifiés10. D’autres approches qui peuvent garantir une incorporation transgène efficace et des taux de survie plus élevés de zygotes injectés ont été étudiées.
Le déterminant clé de l’expression transgène stable et de la transmission à la progéniture est son intégration dans le génome des cellules hôtes. Les lentivirus (VBL) ont la particularité d’être en mesure d’infecter à la fois les cellules de division et de non-division. Leur utilisation comme outil pour l’incorporation de gènes hétérologues dans les embryons s’est avérée très efficace11, et les individus transgéniques sont caractérisés par l’expression stable du fragment d’ADN incorporé. L’efficacité des vecteurs lentiviraux a été confirmée pour la modification génétique des souris12,13, rats12,14, et d’autres espèces11. Dans cette méthode, la suspension LV est injectée sous la zona pellucida de l’embryon au stade de deux pronuclées. Cette technique garantit essentiellement la survie à 100% des embryons parce que l’olémma reste inchangée. La production de suspensions LV de haute qualité et relativement fortement concentrées sont des facteurs cruciaux. Cependant, des concentrations plus faibles de suspensions de VL peuvent être surmontées par des injections répétées11, ce qui augmente la quantité de particules virales à la surface de l’œuf tout en n’affectant pas l’intégration de membrane. Les embryons qui sont soumis à des injections répétées dans l’espace périvitelline se développent davantage, et la progéniture transgénique peut transmettre le transgène par la lignée germinale. L’efficacité de la production de rats transgéniques par transgenèse lentivirale peut atteindre 80 %12.
Ici, nous décrivons la production du lentivirus recombiné dérivé du VIH-1 qui a été pseudotypé avec la protéine d’enveloppe de virus de stomatite vésiculaire (VSV) G. L’utilisation du pseudotype VSV de deuxième génération détermine la grande infectiosité des particules virales et permet la production de vecteurs très stables qui peuvent être concentrés par ultracentrifugation et cryoconservé. Après vérification de titer, les vecteurs sont prêts à être utilisés comme véhicule pour la livraison transgène dans les zygotes de rat Wistar albinos. Après une série d’injections, les embryons peuvent être cultivés du jour au lendemain et transférés à l’étape des deux cellules pour favoriser les mères. À ce stade, l’une des deux approches alternatives peut être envisagée. La procédure standard utilise les femelles pseudo-enceintes comme receveurs d’embryons. Cependant, lorsque le taux de grossesse est faible après l’accouplement avec des mâles vasectomisés, les embryons peuvent être implantés dans les femelles Enceintes Wistar/Sprague-Dawley (SD) qui sont accouplées avec des rats mâles fertiles avec une couleur de fourrure foncée (par exemple, Brown Norway [BN] rats). La couleur de la fourrure permet la distinction de la progéniture de la grossesse naturelle de la progéniture qui proviennent des embryons manipulés transférés.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
La production et l’application de vecteurs viraux étaient conformes aux lignes directrices du niveau 2 de la biosécurité et ont été approuvées par le ministère polonais de l’Environnement. Toutes les procédures expérimentales relatives aux animaux décrites ci-dessous ont été approuvées par le Comité local d’éthique. Les animaux étaient logés dans des cages ventilées individuellement à une température stable (21 à 23 oC) et dans une humidité (50 à 60 %) avec un accès ad libitum à l’eau et à la nourriture dans un cycle clair/obscurité de 12 h/12 h.
1. Production vectorielle lentivirale
- Transfection des cellules HEK 293T
REMARQUE : Le protocole présenté ici est conçu pour la transfection de vingt plats de culture de 10 cm qui produit environ 200 ml de vecteur brut supernatant.- Cellules heK 293T de culture dans le milieu de DMEM qui est complétée avec le sérum bovin foetal (10%, v/v) dans un incubateur humidifié de CO2 à 37 oC. Pour la transfection, préparer vingt plaques de 10 cm de diamètre et les graines de 1,5 à 2 x 106 cellules HEK 293T par plat.
- Lorsque la confluence atteint 70 %, transfectez les cellules à l’aide d’un réactif en polyéthylénimine (PEI), pH 7,0, à un rapport de 3 g de l’Ile-du-Prince-Édouard par 1 g d’ADN.
- Préparer le mélange transfection pour cinq assiettes (préparer le nombre de répétitions en fonction du nombre total de plats). À 1 mL du Medium Aigle modifié (DMEM) de Dulbecco, ajoutez le mélange de trois plasmides afin qu’ils atteignent une quantité finale de 25 g de plasmid VSVg, 50 g de delta R8.2 et 50 g de plasmide codant.
- Pipette de haut en bas, et ajouter 125 l’île de l’Ile-du-Prince-Édouard à une concentration de 3 g/L. Incubate à température ambiante pendant 15 min, en inversant le tube trois fois pendant l’incubation. Ajouter 200 L du mélange transfection par assiette. Ensuite, incuber les plaques dans un incubateur de CO2 humidifié à 37 oC.
- Concentration de vecteurs lentiviraux
- Quarante-huit heures après la transfection, récoltez le milieu qui contient des particules de VBL. Utilisez des tubes coniques de 50 ml.
REMARQUE : Lors de l’utilisation d’un plasmide avec une étiquette fluorescente, les cellules peuvent être visualisées à ce stade pour vérifier l’efficacité de la transfection. Une nouvelle partie du milieu DMEM peut être ajoutée, et les cellules peuvent être incubées pour un supplément de 24 h. Le rendement LV est comparable lorsqu’il est collecté aux points de temps de 48 et 72 h après la transfection. - Centrifuge le milieu à 3.000 x g pendant 5 min et la température ambiante pour enlever les cellules détachées.
- Filtrer le supernatant (0,45 m) et le verser dans de nouveaux tubes.
REMARQUE : Cette étape peut être omise. - Ajouter DNase I (sans RNase, 1 g/mL) et MgCl2 (1 mM), et incuber dans un bain d’eau à 37 oC pendant 15 min.
- Transférer les tubes de polyéthylène moyen à jetable, et l’ultracentrifugeuse dans un rotor oscillant à 115 000 x g et à 4 oC pour 1,5 h.
- Après centrifugage, égouttez doucement les parois des tubes des résidus moyens.
- Faire tremper la granule avec du salin stérile tamponné par phosphate (PBS; 70 à 80 ll par tube).
- Incuber pendant 2 h à 4 à 8 oC.
- Réutilisez les vecteurs viraux dans PBS en passant doucement à la pipe.
CAUTION: Évitez la mousse. - Transférer à un tube de centrifugeuse de 1,5 mL et une centrifugeuse à 7 000 x g et 4 oC pour 30 s. Transférer le supernatant dans un nouveau tube. Répétez cette étape jusqu’à ce qu’aucune granule de débris cellulaires ne soit visible.
- Aliquot et congeler à -80 oC. Évitez de recongeler l’aliquot LV.
- Quarante-huit heures après la transfection, récoltez le milieu qui contient des particules de VBL. Utilisez des tubes coniques de 50 ml.
- Détermination du virus titer à l’aide d’une réaction quantitative en chaîne de polymérase
REMARQUE : La titration des vecteurs viraux est effectuée à l’aide de PCR quantitatif (QPCR). Cette méthode est basée sur l’amplification d’un fragment d’ADN de 84 bp de long à double brin dans la longue région terminale de répétition du génome viral15.- Préparer la courbe standard en faisant des dilutions en série du plasmide de codage LV : 1:500, 1:1,000, 1:5,000, 1:10,000, 1:100,000, et 1:1,000.000. Déterminez le nombre d’exemplaires du plasmide qui est utilisé pour la courbe standard. Utilisez la formule suivante : nombre d’copies/L (concentration [g/L] x 6,02 x 1023 [nombre/mol]) / (660 [g/mol] x plasmid taille [bp]), où 6,02 x 1023 nombre/mol est le numéro d’Avogadro, et 660 g/mol est le poids de bp.
REMARQUE : Des calculatrices de numéros de copie en ligne peuvent être utilisées. - Préparer les dilutions de la suspension lentiviral : 1:100, 1:500 et 1:1,000.
- Préparer le mélange de réaction (volumes par puits) : 10 l de QPCR Mastermix, 1 ll de l’apprêt avant de 10 M, 1 l de l’amorce inversée de 10 M et 7 L de H2O. Pipette le mélange dans les puits de 96 plaques bien.
REMARQUE : Apprêt avant : 5'-AGCTTGCCTTGAGTGCTTCA. Apprêt inversé: 5'-TGACTAAAAGGGTCTGAGGGA. - Ajouter 1 l de chaque dilution standard et suspension lentiviral en triplicate.
- Exécuter le QPCR selon les paramètres suivants : 50 oC pour 2 min, 96 oC pour 5 min, et 35 cycles de 96 oC pour 20 s, 60 oC pour 40 s et 70 oC pendant 1 min, suivis de l’étape de la courbe de fonte : 95 oC pour 1 min et 60 c à 30 s.
- Analyser les résultats en comparant le nombre de molécules reçues pour chaque dilution à la courbe standard. Déterminer la concentration de molécules vectorielles que la moyenne de trois réplique pour chaque dilution.
REMARQUE : La quantification présentée donne la concentration physique des particules virales. Il ne doit pas être traité comme un titer fonctionnel.
- Préparer la courbe standard en faisant des dilutions en série du plasmide de codage LV : 1:500, 1:1,000, 1:5,000, 1:10,000, 1:100,000, et 1:1,000.000. Déterminez le nombre d’exemplaires du plasmide qui est utilisé pour la courbe standard. Utilisez la formule suivante : nombre d’copies/L (concentration [g/L] x 6,02 x 1023 [nombre/mol]) / (660 [g/mol] x plasmid taille [bp]), où 6,02 x 1023 nombre/mol est le numéro d’Avogadro, et 660 g/mol est le poids de bp.
2. Génération de rats transgéniques
- Superovulation et collecte d’embryons fécondés
- Administrer des gonadotropines.
REMARQUE : Pour augmenter le nombre d’embryons recueillis (environ 30 par femelle), utilisez des femelles Wistar immatures de 5 semaines pour la stimulation hormonale.- Le jour 1 (12 PM-1 PM), injectez intraperitoneally la gonadotropine sérique de la jument enceinte (PMSG ; 25 UI par femelle). Préparer 1 mL aliquots de solution de travail à une concentration de 125 UI/mL en dissolvant la poudre d’hormone dans 0,9% NaCl. Conserver à -20 oC jusqu’à 1 mois ou -80 oC pour un temps allant jusqu’à 6 mois.
- Le jour 3 (12 PM-1 PM), injectez intraperitoneally la gonadotrophine chorionique humaine (hCG ; 30 UI par femelle). Préparer 1 mL aliquots de solution de travail (150 UI/mL) en dissolvant la poudre d’hormone dans 0,9% NaCl. Conserver à -20 oC jusqu’à 1 mois ou -80 oC pour un temps allant jusqu’à 6 mois.
- Après l’administration de hCG, s’accouplent les femelles 1:1 avec des mâles sexuellement fertiles (3-10 mois).
- Le lendemain matin (jour 4 à 8-10 AM), vérifiez les femelles pour la présence d’une prise vaginale. Vérifiez l’ouverture vaginale pour la présence d’une prise d’accouplement blanchâtre, qui pour la meilleure visualisation devrait être vérifiée tôt le matin après la nuit d’accouplement. Pour la collecte d’embryons, n’utilisez que les femelles avec une prise visible.
- Recueillir des embryons à 10 heures. Sacrifiez les animaux pour exciser les oviductes et recueillir les oviductes dans un plat avec un support M2 pré-réchauffé.
- Transférer les oviductes dans un plat de 35 mm qui contient un milieu M2 préchauffé avec de l’hyaluronidase des testicules bovins à une concentration de 0,5 mg/mL.
- Ouvrez les parois de l’oviduct à l’aide de forceps fins sous un stéréomicroscope et appuyez sur l’ampulla (c’est-à-dire la partie gonflée de l’oviduct qui contient des embryons fécondés qui sont entourés de cellules cumulus) jusqu’à ce que les embryons soient libérés.
REMARQUE : Hyaluronidase digère enzymatiquement les cellules de cumulus, libérant des embryons.
CAUTION : L’exposition prolongée à l’hyaluronidase est nocive pour les embryons ; par conséquent, cette étape ne devrait pas durer plus de 5 min. - Pour faciliter la libération d’embryons provenant de cellules cumulus, les pipette doucement de haut en bas à l’aide d’une pipette de transfert de verre qui est reliée à un tube d’aspirateur actionné par la bouche.
- Pour produire la pipette de transfert, tirez une pipette Pasteur en verre sur une flamme pour produire une pointe droite de 5 à 10 cm. Casser la pipette en laissant une pointe de 4 cm.
- Laver les embryons à quelques reprises dans le milieu M2 pour enlever l’hyaluronidase et les débris cellulaires. Transférer les embryons dans un plat de 60 mm qui contient des gouttes de M16 pré-équilibrés, recouvertes de paraffine liquide ou d’huile minérale, dans un incubateur humidifié de 37 oC avec une atmosphère deCO 2 de 5 %.
- Administrer des gonadotropines.
- Microinjection de vecteurs lentiviraux à l’embryon à un étage sous la zona pellucida
REMARQUE : Utilisez des embryons à une cellule avec deux pronuclées visibles pour la microinjection(figure 1).- Décongeler l’aliquot LV à température ambiante et la centrifugeuse à 10 000 x g et RT pendant 2 min pour pelleter les débris cellulaires restants.
- Configuration de micro-injection
- Préparer les pipettes en verre (capillaire en verre borosilicate) à l’aide d’une microforge. Tirez le capillaire de verre sur une flamme pour produire une pointe de 5 à 10 cm. Casser la pipette en laissant une pointe de 4 cm. Le diamètre extérieur doit être de 80 à 120 m.
REMARQUE : Assurez-vous que la pointe de la pipette est parfaitement droite et lisse. - Assembler la pipette tirée dans une microforge avec la pointe devant le filament de chauffage. Chauffer le filament très près de la pointe de la pipette et lui permettre de rétrécir à un diamètre de 15 m (environ 20 % de la taille de l’embryon). Placez la pipette perpendiculairement au filament chauffant, de 2 à 3 mm de la pointe de la pipette, et commencez à chauffer. Le verre ramollira. Chauffer jusqu’à ce qu’il atteigne un angle de 15 degrés.
- Préparer les capillaires en verre de borosilicate de microinjection avec un filament à l’aide d’un puller pipette. Insérer le capillaire dans la chambre de traction. Exécuter un essai de rampe (pour la première fois pour le nouveau verre et à chaque fois après avoir changé le filament). Réglez la chaleur à la valeur de la rampe -10, Pull à 100, Velocity à 150 et Time to 100.
REMARQUE : Modifier les paramètres pour obtenir un capillaire d’injection optimal. - Sous un capot de flux lamineur biosécurité, chargez environ 2 lil de la solution virale dans la pipette de microinjection avec une pointe de microchargeur.
- Préparer un plat de microinjection (couvercle de 60 mm de plat Petri) avec une goutte de 100 l de M2 moyen (au milieu), recouverte de paraffine liquide ou d’huile minérale.
- Montez la pipette de fixation et le capillaire de microinjection qui est chargé avec la solution virale à un micromanipulateur et un plat de microinjection sous un microscope inversé.
- Préparer les pipettes en verre (capillaire en verre borosilicate) à l’aide d’une microforge. Tirez le capillaire de verre sur une flamme pour produire une pointe de 5 à 10 cm. Casser la pipette en laissant une pointe de 4 cm. Le diamètre extérieur doit être de 80 à 120 m.
- Effectuez la microinjection.
- Transférer 15 à 20 embryons à un étage sur la chute M2 sur le plat de microinjection. Tenez l’embryon à l’aide d’une pipette de retenue.
- À l’aide du grossissement 400x, injectez la solution LV sous la zona pellucida à l’espace périvitelline à l’aide du capillaire en verre qui est connecté à un injecteur automatique. Tenez le capillaire sous la zona pellucida pendant un moment.
REMARQUE : En utilisant une légère pression positive, la solution virale s’écoulera continuellement hors du capillaire d’injection, mais le volume de la suspension qui est livrée ne peut pas être contrôlé. - À l’aide d’une pipette fine, remettre les embryons au plat de culture de l’incubateur à 37 oC dans une atmosphère de CO2 de 5 %. Le nombre d’injections d’un zygote peut varier et peut être adapté en fonction de la concentration virale de vecteur.
REMARQUE : Les embryons injectés peuvent être transférés à des mères adoptives au stade d’une cellule ou à un milieu O/N incubé dans le milieu M16 avant d’être transférés au stade à deux cellules. La culture in vitro prolongée des embryons de rats doit être évitée.
- Transfert d’embryons injectés aux mères adoptives
- Préparez les mères adoptives en accouplant des femelles SD sexuellement matures avec des mâles BN fertiles ou avec des mâles SD vasectomisés (la procédure de vasectomie est décrite à la section 3 ci-dessous) le jour 3 (pour le transfert d’embryons au stade d’une cellule) ou au quatrième jour (pour le transfert d’embryons au stade à deux cellules).
REMARQUE : Pour le transfert d’oviduct, utilisez 0.5 jours après coitum (dpc) femelles. - Le lendemain matin, vérifiez les femelles SD pour une prise vaginale, et n’utilisez que ceux avec une prise visible.
- Effectuez le transfert d’embryons.
REMARQUE : Conduisez la procédure chirurgicale avec des instruments stériles sous un stéréomicroscope. Avant le jour de la chirurgie, ciseaux autoclave, forceps fins, porte-aiguille, et porte-scalpel.- Anesthésiez une femelle avec l’administration i.p. de la kétamine (50 mg/kg) et de la médénodine (0,5 mg/kg). Testez les réflexes pour confirmer l’anesthésie avant de commencer l’intervention chirurgicale.
- Injecter l’animal sous-cutanée avec de l’acide toléfenamique (2 mg/kg), du tartrate de butorphanol (1 mg/kg) et de l’enrofloksacine (5 à 10 mg/kg) pour prévenir l’inflammation, la douleur et l’infection, respectivement.
- Appliquer la lubrification de la pommade ophtalmique aux deux yeux pour prévenir le séchage cornéen. Rasez la fourrure du dos et stérilisez la peau avec un gommage chirurgical suivi de 70 % d’alcool à l’aide de tampons stériles non adhérants. Laisser sécher la peau.
- Injecter l’animal sous-cutanée avec 100 L de bupivacaine de 0,25% (anesthésique local) au site d’incision. Transférer l’animal en position couchée sur une surface propre sur un coussin chauffant dans le but d’un microscope chirurgical. Couvrir le rat d’un drapé stérile d’un petit trou coupé sur le bas du dos.
- Effectuer une incision cutanée d’environ 2 cm, parallèle à la colonne vertébrale lombaire.
- À l’aide de ciseaux pointus, faire une coupure dans la paroi abdominale. Prenez un coussin de graisse ovarienne à l’aide de forceps, et retirez l’ovaire et l’oviducte et placez-les sur de la gaze qui est mouillée avec 0,9% NaCl.
- Aspirer le milieu M2, trois bulles d’air et les embryons dans le capillaire de transfert. Nombre total recommandé d’embryons à transférer (unilatérales ou bilatérales) : femelle enceinte (embryons de 15 à 16 ans), pseudo-femelles (30 embryons).
- Faire une petite incision dans l’oviducte (entre l’infundibulum et l’ampulla) à l’aide de micro-ciseaux, et insérer la pipette de transfert dans l’oviducte.
- Expulser délicatement les embryons et les bulles d’air de la pipette vers l’oviducte. Avec des forceps émoussés, placez l’appareil reproducteur dans la cavité abdominale.
- Suture de la paroi abdominale avec de l’acide polyglycolique sutures absorbables et fermer l’incision de la peau avec des pinces de plaie. Selon le nombre d’embryons disponibles, répétez cette procédure pour l’autre oviducte.
- Injecter l’animal intraperitoneally avec l’atipamezole (0,5 mg/kg) pour inverser l’effet de l’anesthésie.
- Transférer l’animal dans une cage propre et le garder sur une plaque chauffante pour récupérer complètement de l’anesthésie. La livraison chez les rats se produit après 21 jours.
REMARQUE : Lorsque les rats mâles BN sont utilisés pour l’accouplement, seuls les chiots blancs sont potentiellement transgéniques; les chiots bruns sont de grossesse naturelle. - Recueillir des fragments de tissu (de préférence de l’oreille) au génotype de 3 semaines.
- Préparez les mères adoptives en accouplant des femelles SD sexuellement matures avec des mâles BN fertiles ou avec des mâles SD vasectomisés (la procédure de vasectomie est décrite à la section 3 ci-dessous) le jour 3 (pour le transfert d’embryons au stade d’une cellule) ou au quatrième jour (pour le transfert d’embryons au stade à deux cellules).
3. Vasectomie
REMARQUE : Avant le jour de la chirurgie, ciseaux autoclave, forceps fins et porte-aiguilles.
- Anesthésiez un rat SD mâle de 5 semaines avec l’administration i.p. de la kétamine (50 mg/kg) et de la médénodine (0,5 mg/kg). Testez les réflexes pour confirmer l’anesthésie avant de commencer l’intervention chirurgicale.
- Administrer de l’acide tolfenamic (2 mg/kg), du tartrate de butorphanol (1 mg/kg) et de l’enrofloksacine (5 à 10 mg/kg) sous-cutanée pour prévenir l’inflammation, la douleur et l’infection, respectivement.
- Appliquer la lubrification de la pommade ophtalmique aux deux yeux pour prévenir le séchage cornéen. Placez le rat supine sur une surface propre sur un coussin chauffant, et stérilisez la peau sur les testicules avec des gommage chirurgicaux suivis de 70% d’alcool à l’aide de tampons stériles non adhérants. Laisser sécher la peau. Couvrir le rat d’un drapé stérile d’un petit trou coupé sur les testicules. Appuyez doucement sur l’abdomen pour exposer les testicules dans le sac scrotal.
- À l’aide de ciseaux chirurgicaux, faites une incision de 0,5 cm au milieu du sac scrotal. Localiser le mur de ligne médiane (ligne blanchâtre) entre les testicules.
- Faire une incision de 5 mm dans la membrane des testicules près du côté gauche de la paroi de la ligne médiane.
- Poussez soigneusement les testicules vers la gauche et localisez les différenciations vas (entre les testicules et la ligne médiane) comme un conduit blanc avec un seul vaisseau sanguin.
- Tirez doucement les vas deferens hors du sac scrotal à l’aide des forceps d’un horloger. Tenir le vas différencié d’une paire de forceps, et couper avec des ciseaux fins (ou cautériser avec des pointes rouges d’une deuxième paire de forceps). Retirer un fragment de 1 cm du conduit.
REMARQUE : Si la cautérisation est effectuée, maintenez la pointe de la deuxième paire de forceps dans la flamme. - Répétez la procédure ci-dessus pour les autres testicules. Suture de la peau avec de l’acide polyglycolique sutures absorbables et injecter l’animal intraperitoneally avec de l’atipamezole (0,5 mg/kg).
- Placez le rat dans une cage propre sur une plaque chauffante jusqu’à ce que l’animal se rétablisse de l’anesthésie.
REMARQUE : Les mâles peuvent être utilisés dans les accouplements d’essai après une période de récupération de 2 semaines. Une fois la stérilité confirmée, ils peuvent être utilisés pour l’induction pseudo-grossesse.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
À l’aide du protocole décrit dans les présentes, des vecteurs lentiviraux qui transportaient la construction Syn-TDP-43-eGFP ont été produits (3,4 x 108/L) et pourraient ensuite être utilisés pour des injections subzonales embryonnaires à un stade cellulaire. Seuls les embryons avec deux pronuclées visibles ont été soumis à la procédure. Le nombre d’injections de suspensions virales a été déterminé expérimentalement. Une efficacité élevée d’implantation et un manque simultané de progéniture transgénique ont été considérés comme des indicateurs d’un nombre insuffisant de particules virales pour la transduction réussie. Dans ce cas, le nombre d’injections a été augmenté. L’administration unique de LV a eu comme conséquence la naissance de rats de 20 F0-génération, aucun d’entre eux étaient transgéniques. Une augmentation du nombre d’injections d’un ordre de grandeur n’a pas entraîné la naissance de rats, mais 100% des embryons se sont développés au stade à deux cellules. Dans les expériences suivantes, le nombre d’injections a été augmenté d’un par rapport à la valeur pour laquelle la progéniture a été obtenue. Pour la variante de deux injections, huit rats sont nés, dont trois ont été confirmés pour porter le transgène (résumé dans le tableau 1). L’un des fondateurs n’a pas transféré le transgène à sa progéniture. Le nombre d’embryons qui ont été injectés et transférés dans chaque variante expérimentale était de 48 dans les variantes LV x1 et LV x2 et 45 dans LV x10. Trois femelles d’accueil ont été employées pour chaque installation expérimentale. L’approche choisie a permis la génération de lignées de rats transgéniques stables qui ont exprimé la protéine de fusion TDP-43-eGFP sous contrôle du promoteur neuronal Synapsin-1 dans l’ensemble du système nerveux central(figure 2A,B)14. La transgenèse à base de Lentivirus a donné lieu à une seule copie d’insertion du transgène comme le démontre qPCR(figure 2C).
Dans la configuration expérimentale décrite ci-dessus, le taux de survie des embryons injectés était de 95%. Des résultats similaires ont été obtenus lorsque la même méthode a été utilisée pour d’autres vecteurs lentiviraux comme résumé dans le tableau 2. Le pourcentage d’embryons qui ont survécu aux injections pronucléaires était significativement plus faible (29-45%). Le tableau 2 résume les résultats représentatifs de l’efficacité d’implantation des zygotes manipulés, compte tenu du transfert de pseudo-femmes enceintes. L’utilisation d’embryons non manipulés ainsi que des embryons injectés a déjà été signalée16. Nos résultats globaux suggèrent que les rats femelles enceintes peuvent être employés comme mères d’accueil avec l’efficacité comparable. Nous avons obtenu un pourcentage similaire d’implantation d’embryons étrangers chez des rats enceintes et pseudo-étudiants (moyenne globale pour plusieurs configurations expérimentales : 15 % contre 16 %). Cependant, le taux d’implantation était plus élevé lorsque les embryons ont subi une manipulation plus subtile, ce qui signifie une injection subzonale (10% contre 21%). Notamment, les données numériques qui ont été analysées pour des séries individuelles de microinjection ont indiqué que l’efficacité de l’implantation dépendait du nombre d’injections d’un embryon(tableau 1,dernière colonne) et dépendait indirectement de la charge virale.
| Vecteur | nombre d’injections/embryons | nombre d’embryons injectés | nombre de chiots | nombre de mères adoptives | nombre de fondateurs transgéniques | Efficacité d’implantation pour chaque variante |
| Syn-TDP-43WTLV | 1 | 48 | 20 | 3 | 0 | 42% |
| 10 | 45 | 0 | 3 | 0 | 0% | |
| 2 | 48 | 8 | 3 | 3 | 17% |
Tableau 1 : Résumé du nombre d’injections sous-zonales de zygotes avecdes vecteurs lentiviraux Syn-TDP-43WT.
| Méthode | Vecteur | Titer/ Concentration | Nombre d’embryons injectés | Embryons survécus | Taux de survie | Nombre de mères d’accueil | Nombre de chiots | Efficacité de l’implantation | Grossesse (P) /Pseudopregnancy (PP) |
| Pni | TTYH1-Thy1-EGFP | 1 ng/L | 1083 | 424 | 39% | 16 | 54 | 13% | Pp |
| Pni | H3mCherry (H3mCherry) | 0,5-2 ng/L | 2229 | 647 | 29% | 29 | 67 | 10% | Pp |
| Pni | Syn-TDP-43-A315T | 2 ng/L | 1256 | 562 | 45% | 31 | 42 | 7% | Pp |
| Lv | Syn-TDP-43-A315T | 8,7 x 108 | 115 | 106 | 92% | 7 | 18 | 17% | P |
| Lv | Syn-TDP-43 WT | 3,4 x 108 | 152 | 141 | 93% | 9 | 28 | 20% | P |
| Lv | LVH3mcherry | 1,3 x 107 | 504 | 450 | 89% | 13 | 115 | 26% | Pp |
Tableau 2 : Taux de survie et efficacité d’implantation de l’embryon, selon la méthode d’injection utilisée et la grossesse par rapport à l’induction pseudo-grossesse. PNI, injection pronucléaire; LV, l’injection sous-zonale vectorielle lentiviral.

Figure 1 : Photographie microscopique de l’embryon de rat à un étage à une cellule qui a été préparé pour l’injection vectorielle lentivirale subzonale. L’embryon a été immobilisé avec une pipette de fixation. Deux pronuclées qui contenaient du matériel génétique maternel et paternel et le corps polaire sont visibles. Barre d’échelle à 20 m. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
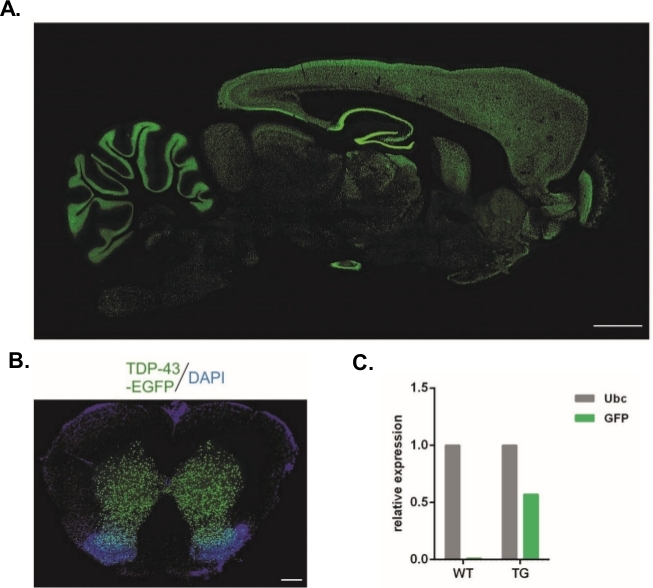
Figure 2 : Génération de lignées de rats transgéniques stables qui exprimaient la protéine de fusion TDP-43-eGFP sous contrôle du promoteur neuronal Synapsin-1 dans tout le système nerveux central. (A) Synapsin-1 (Syn)-conduit hTDP-43-eGFP modèle d’expression dans une section sagittale du cerveau transgénique de rat. Barre d’échelle de 3 mm. (B) Section coronale de la moelle épinière d’un rat transgénique où la fluorescence eGFP, contre-limitée au DAPI, était limitée à la matière grise de la moelle épinière. Barre d’échelle à 250 m. (C) Expression relative de la transcription transgène de GFP comparée à la transcription de référence d’ubiquitin C. n 2 wildtype. n 2 transgéniques. Le chiffre a été modifié à partir de14. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Les progrès des technologies transgéniques ont fait des modèles de rongeurs un outil inestimable dans la recherche biomédicale. Ils offrent l’occasion d’étudier les relations génotype-phénotype in vivo. Ici, nous présentons une alternative largement disponible pour la transgenèse conventionnelle par des injections pronucléaires. L’utilisation de la transduction de gène lentiviral contourne la nécessité d’exiger des microinjections parce que des vecteurs viraux peuvent être injectés sous la zona pellucida. Cette approche n’affecte pas l’intégrité de l’embryon, qui garantit essentiellement un taux de survie de 100 % pour les zygotes injectés. Le transgène qui est incorporé au moyen de vecteurs lentiviraux est intégré de façon stable dans le génome hôte, permettant l’expression à long terme et la transmission germinale. En outre, nous présentons deux techniques alternatives pour le transfert d’embryons modifiés aux mères adoptives. Une technique utilise le transfert d’embryons aux femelles pseudo-enceintes qui sont précédemment préparées en s’accouplant avec des mâles infertiles vasectomisés. L’autre technique est basée sur l’utilisation de femelles naturellement enceintes qui sont accouplées avec des mâles fertiles, mais avec une couleur de fourrure différente (c.-à-d., les rats BN). Ce cours plus physiologique de la grossesse permet le bon développement d’embryons qui subissent des modifications génétiques difficiles16.
Les premières tentatives réussies pour générer des rats transgéniques ont été rapportées en 19907. Cependant, en raison des difficultés dans la transgenèse de rat17, un nombre relativement faible de lignées de rat transgéniques ont été générées au cours des dernières décennies9. Plusieurs différences principales sont observées entre la transgenèse de souris et de rat utilisant des microinjections. Pour les rats, les lignées principalement dépassées (p. ex., Wistar et SD) sont utilisées pour la transgenèse. Pour les souris, les chercheurs utilisent principalement le croisement F1 des souches consanguines en raison de leur fertilité plus élevée, une meilleure réponse à la superovulation hormonale, et le développement relativement facile des embryons in vitro du stade à une cellule aux blastocystes18. L’induction de la superovulation chez les rats est beaucoup moins efficace que chez les souris utilisant la stimulation standard de l’hormone PMSG/hCG. Pour cette raison, des tentatives ont été faites pour développer d’autres protocoles pour administrer ces hormones chez les rats qui utilisent l’infusion continue de FSH au lieu d’une administration PMSGunique 19. Cependant, la superovulation qui est causée par PMSG/hCG ou FSH/hCG a été montré pour avoir une efficacité comparable20. À notre avis, le facteur le plus critique qui affecte l’efficacité de la superovulation est l’âge des femmes sélectionnées. Néanmoins, les paramètres exacts doivent être testés pour chaque souche de rat, laboratoire, etc.
La procédure d’injection de solution d’ADN dans le pronucléus d’un embryon unicellulaire est similaire pour les deux espèces de rongeurs. Cependant, les pronuclées des zygotes de rat n’ont pas des formes si régulières que chez les souris et ont tendance à être plus difficiles à définir dans le cytoplasme de la cellule. En outre, la membrane de cellules de zygote de rat et la membrane pronucléaire sont plus élastiques et visqueuses, compliquant ainsi l’insertion d’une micropipette de verre qui est chargée avec la solution d’ADN. Ces facteurs conduisent à des taux de survie aux œufs de rat plus faibles après la microinjection (31-65% contre 80% chez la souris) et expliquent l’efficacité plus faible de transgenèse chez les rats9. En outre, une manipulation intensive et mécanique de l’embryon peut également affecter l’efficacité de l’implantation, qui dans de nombreux laboratoires, y compris le nôtre, atteint un maximum de 10%. Ce rendement relativement faible est observé même après l’implantation d’un nombre approprié d’embryons21.
Une méthode qui surmonte les difficultés susmentionnées est l’infection des embryons unicellulaires avec des rétrovirus. Les rétrovirus contiennent du matériel génétique sous forme d’ARN, qui, à l’entrée dans la cellule infectée, est transcrit dans l’ADN par transcriptase inverse du virus. L’ADN est ensuite transporté à travers les pores nucléaires vers le noyau cellulaire, où il s’intègre dans le génome de la cellule sous la forme d’un provirus. Des vecteurs lentiviraux ont été utilisés pour générer des souris et des rats transgéniques12,14,22. Les embryons unicellulaires qui n’ont pas de zona pellucida peuvent être incubés dans une solution avec un vecteur lentiviral, ou le vecteur peut être injecté sous la zona pellucida dans l’espace périvitelline. Le principal avantage de cette méthode est son efficacité extrêmement élevée, atteignant plus de 80% de la progéniture transgénique. Après l’infection par le vecteur lentiviral, de nombreuses copies à différents sites peuvent s’intégrer dans le génome du zygote, contrairement à la méthode de transgenèse par microinjection pronucléaire, dans laquelle un site d’intégration est généralement observé12. Dans la progéniture du fondateur transgénique qui est faite à l’aide de vecteurs lentiviraux, des copies individuelles du transgène sont séparées, qui peuvent être manifestées par différents profils d’expression du transgène dans chacune de la progéniture. Cependant, cela peut augmenter le risque de recevoir un sujet avec le profil d’expression souhaité qui est dérivé du transgène. Les restrictions s’appliquent principalement à la taille du transgène, qui est limité à environ 8 kb23.
Une autre difficulté dans la transgenèse de rat est la génération de femelles qui servent de mères porteuses pour les embryons génétiquement modifiés. Dans la procédure standard, les femelles sont croisées avec des mâles vasectomisés stériles pour induire la pseudoprégnance. Chez les rats, la technique d’évaluation de la pseudoprégnance est beaucoup plus difficile que chez la souris, de sorte que la stimulation avec le gonadotropin libérant l’agoniste d’hormone est parfois employée quelques jours avant l’accouplement avec des mâles. Pour ces raisons, dans le protocole décrit, nous offrons deux approches alternatives pour obtenir des mères adoptives. L’efficacité globale d’implantation des zygotes manipulés lorsque des femelles enceintes ou pseudo-enceintes sont utilisées est similaire. Cependant, la présence d’embryons naturels et non manipulés ainsi que des embryons manipulés peut améliorer le taux de grossesse16. Bien que la principale différence dans le taux d’implantation soit la technique de manipulation (c.-à-d. PNI vs LV, 10 % contre 20 %; voir le tableau 2), l’utilisation de femelles pseudo-enceintes comme mères adoptives peut être bénéfique pour certaines expériences.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
L’auteur (W.K.) a droit au brevet, "Méthode de production d’un animal transgénique", de l’office des brevets de la République de Pologne (no 355353; 21.03.2008).
Acknowledgments
Cette étude a été soutenue par le projet ANIMOD dans le cadre du programme Team Tech Core Facility Plus de la Fondation pour la science polonaise, cofinancé par l’Union européenne dans le cadre du Fonds européen de développement régional à WK.
Materials
| Name | Company | Catalog Number | Comments |
| 7500 Real Time PCR System | Applied Biosystems | ||
| Aerrane (isoflurane) | Baxter | FDG9623 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177-5EA | |
| Atipam 5 mg/ml | Eurovet Animal Health BV | N/A | 0.5 mg/kg |
| Baytril 25 mg/ml (enrofloksacin) | Bayer | N/A | 5-10 mg/kg |
| Borosilicate glass capillaries with filament GC100TF-15 | Harvard Apparatus Limited | 30-0039 | injection capillary |
| Bupivacaine 25 mg/ml | Advanz Pharma | N/A | 0.25% in 0.9% NaCl |
| Butomidor 10 mg/ml (butorphanol tartrate) | Orion Pharma | N/A | 1 mg/kg |
| CELLSTAR Tissue Cell Culture Dish 35-mm | Greiner Bio-One | 627160 | |
| CELLSTAR Tissue Cell Culture Dish 60-mm | Greiner Bio-One | 628160 | |
| CellTram Oil | Eppendorf | 5176 000.025 | |
| Cepetor (Medetomidine) 1 mg/ml | cp-pharma | N/A | 0.5 mg/kg |
| Chorulon, Human Chorionic Gonadotrophin | Intervet | N/A | 150 IU/ ml ml 0.9% NaCl |
| DMEM low glucose | Sigma Aldrich | D6048 | |
| DNase, RNase-free | A&A Biotechnology | 1009-100 | |
| EmbryoMax Filtered Light Mineral Oil | Sigma | ES-005-C | |
| Envelope protein coding plasmid for lentiviral vectors (VSVg plasmid) | ADDGENE | 14888 | |
| FemtoJet | Eppendorf | 4i /5252 000.013 | |
| Fetal Bovine Serum | Sigma Aldrich | F9665-500ML | |
| Folligon, Pregnant Mare’s Serum Gonadotropin | Intervet | N/A | 125 IU/ml in .9% NaCl |
| HEK 293T cells | ATCC | ATCC CRL-3216 | |
| Hyaluronidase from Bovine Testis | Sigma | H4272-30MG | 0.5 mg/ml in M2 medium |
| Inverted Microscope | Zeiss | Axiovert 200 | |
| Ketamine 100mg/ml | Biowet Pulawy | N/A | 50 mg/kg |
| Liquid Paraffin | Merck Millipore | 8042-47-5 | |
| M16 medium EmbryoMax | Sigma | MR-016-D | |
| M2 medium | Sigma | M7167 | |
| Magnesium Chloride 1M | Sigma Aldrich | 63069-100ML | |
| Microforge | Narishige | MF-900 | |
| Mineral Oil | Sigma | M8410-500ML | |
| NaCl 0.9% | POLPHARMA OTC | N/A | sterile, 5ml ampules |
| Operation microscope | Inami Ophthalmic Instruments | Deca-21 | |
| Packaging system coding plasmid for lentiviral vectors (delta R8.2 plasmid) | ADDGENE | 12263 | |
| PEI reagent (Polyethylenimine, Mw ~ 25,000,), | Polysciences, Inc | 23966-1 | |
| Penicilin-streptomycin | Sigma Aldrich | P0781-100ML | |
| Phosphate Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma Aldrich | 806552-500ML | |
| Puller | Sutter Instrument Co. | P-97 | |
| Reflex Clip Applier/Reflex Clips | World Precision Instruments | 500345/500346 | |
| Safil, polyglycolic acid, braided, coated, absorbable threads | B.Braun Surgical | 1048029 | |
| Stereomicroscope | Olympus | SZX16 | |
| Surgical Sewing Thread | B.Braun | C1048040 | |
| SYBR Green PCR Master Mix | Applied Biosystem | 4334973 | |
| Tolfedine 4% (tolfenamic acid) | Vetoquinol | N/A | 2 mg/kg |
| TransferMan NK2 | Eppendorf | N/A | |
| Trypsin EDTA solution | Sigma Aldrich | T3924-500ML | |
| Ultracentrifuge | Beckman Coulter | Optima L-100 XP | |
| VacuTip | Eppendorf | 5175108.000 | holders capillary |
| Vita-POS | Ursapharm | N/A | eye ointment |
| Warming Plate | Semic | N/A | |
| Watchmaker Forceps | VWR | 470018-868 |
References
- Lazar, J., Moreno, C., Jacob, H. J., Kwitek, A. E. Impact of genomics on research in the rat. Genome Research. 15 (12), 1717-1728 (2005).
- Tarkowski, A. K. Studies on mouse chimeras developed from eggs fused in vitro. National Cancer Institute Monographs. 11, 51-71 (1963).
- Gordon, J. W., Ruddle, F. H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 214 (4526), 1244-1246 (1981).
- Gill, T. J., Smith, G. J., Wissler, R. W., Kunz, H. W. The Rat as an Experimental Animal. Science. 245 (4915), 269-276 (1989).
- Aitman, T. J., et al. Progress and prospects in rat genetics: a community view. Nature Genetics. 40 (5), 516-522 (2008).
- Hammer, R. E., Maika, S. D., Richardson, J. A., Tang, J. P., Taurog, J. D. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 63 (5), 1099-1112 (1990).
- Mullins, J. J., Peters, J., Ganten, D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 344 (6266), 541-544 (1990).
- Menoret, S., Remy, S., Usal, C., Tesson, L., Anegon, I. Generation of Transgenic Rats by Microinjection of Short DNA Fragments. Rat Genomics: Methods and Protocols. 597, 81-92 (2010).
- Tesson, L., et al.
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: Technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Ritchie, W. A., Neil, C., King, T., Whitelaw, C. B. Transgenic embryos and mice produced from low titre lentiviral vectors. Transgenic Research. 16 (5), 661-664 (2007).
- Lois, C., Hong, E. J., Pease, S., Brown, E. J., Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295 (5556), 868-872 (2002).
- Pfeifer, A., Ikawa, M., Dayn, Y., Verma, I. M. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proceedings of the National Academy of Sciences of the United States of America. 99 (4), 2140-2145 (2002).
- Koza, P., et al. Neuronal TDP-43 depletion affects activity-dependent plasticity. Neurobiology of Disease. 130, 104499 (2019).
- Scherr, M., Battmer, K., Blomer, U., Ganser, A., Grez, M. Quantitative determination of lentiviral vector particle numbers by real-time PCR. Biotechniques. 31 (3), 520 (2001).
- Canseco, R. S., et al. Gene transfer efficiency during gestation and the influence of co-transfer of non-manipulated embryos on production of transgenic mice. Transgenic Research. 3 (1), 20-25 (1994).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Yagle, M. K., Palmiter, R. D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proceedings of the National Academy of Sciences of the United States of America. 82 (13), 4438-4442 (1985).
- Armstrong, D. T., Opavsky, M. A. Superovulation of immature rats by continuous infusion of follicle-stimulating hormone. Biology of Reproduction. 39 (3), 511-518 (1988).
- Popova, E., Krivokharchenko, A., Ganten, D., Bader, M. Comparison between PMSG- and FSH-induced superovulation for the generation of transgenic rats. Molecular Reproduction and Development. 63 (2), 177-182 (2002).
- Johnson, L. W., Moffatt, R. J., Bartol, F. F., Pinkert, C. A. Optimization of embryo transfer protocols for mice. Theriogenology. 46 (7), 1267-1276 (1996).
- van den Brandt, J., Wang, D., Kwon, S. H., Heinkelein, M., Reichardt, H. M. Lentivirally generated eGFP-transgenic rats allow efficient cell tracking in vivo. Genesis. 39 (2), 94-99 (2004).
- Remy, S., et al. The Use of Lentiviral Vectors to Obtain Transgenic Rats. Rat Genomics: Methods and Protocols. 597, 109-125 (2010).
