

Summary
Dieser Artikel zielt darauf ab, die Methodik für die lentivirale Transgenese bei Rattenembryonen unter Verwendung mehrerer Injektionen einer Virussuspension in den zygoten Perivitelline-Raum bereitzustellen. Weibliche Ratten, die mit einem fruchtbaren männlichen Stamm mit einer anderen dominanten Fellfarbe gepaart werden, werden verwendet, um pseudoschwangere Pflegemütter zu erzeugen.
Abstract
Transgene Tiermodelle sind für die moderne biomedizinische Forschung von grundlegender Bedeutung. Die Einbindung fremder Gene in frühe Maus- oder Rattenembryonen ist ein unschätzbares Werkzeug für die Genfunktionsanalyse in lebenden Organismen. Die Standard-Transgenese-Methode basiert auf der Mikroinjektion fremder DNA-Fragmente in einen Pronukle einer befruchteten Oozyte. Diese Technik ist bei Mäusen weit verbreitet, bleibt aber bei anderen Tierarten relativ ineffizient und technisch anspruchsvoll. Das Transgen kann auch über eine Lentiviralinfektion in einzellige Embryonen eingebracht werden, was eine wirksame Alternative zu standardpronuklearen Injektionen darstellt, insbesondere bei Arten oder Stämmen mit einer anspruchsvolleren Embryostruktur. Bei diesem Ansatz wird eine Suspension, die lentivirale Vektoren enthält, in den Perivitelinraum eines befruchteten Rattenembryons injiziert, der technisch weniger anspruchsvoll ist und eine höhere Erfolgsrate hat. Es wurde gezeigt, dass Lentivirale Vektoren das Transgen effizient in das Genom integrieren, um die Erzeugung stabiler transgener Linien zu bestimmen. Trotz einiger Einschränkungen (z. B. Anforderungen an die Biosicherheitsstufe 2, Grenzwerte für die Größe von DNA-Fragmenten) ist die lentivirale Transgenese eine schnelle und effiziente Transgenesemethode. Zusätzlich wird die Verwendung weiblicher Ratten, die mit einem fruchtbaren männlichen Stamm mit einer anderen dominanten Fellfarbe gepaart sind, als Alternative zur Erzeugung pseudoschwangerer Pflegemütter dargestellt.
Introduction
Seit vielen Jahren werden Labornagetiere wie Ratten und Mäuse verwendet, um menschliche physiologische und pathologische Bedingungen zu modellieren. Tierforschung hat zu Entdeckungen geführt, die auf andere Weise unerreichbar waren. Zunächst konzentrierten sich genetische Studien auf die Analyse spontan auftretender Störungen und Phänotypen, die den menschlichenZustandeng nachahmen 1 . Die Entwicklung gentechnischer Methoden ermöglichte die Ein- oder Löschung bestimmter Gene, um einen gewünschten Phänotyp zu erhalten. Daher wird die Erzeugung transgener Tiere als eine grundlegende Technik in der modernen Forschung anerkannt, die Studien über die Genfunktion in lebenden Organismen ermöglicht.
Die transgene Tiertechnologie ist durch eine Kombination von Errungenschaften in der experimentellen Embryologie und Molekularbiologie möglich geworden. In den 1960er Jahren veröffentlichte der polnische Embryologe A. K. Tarkowski die ersten Arbeiten zur Manipulation von Mausembryonen in den frühen Stadien der Entwicklung2. Zusätzlich entwickelten Molekularbiologen Techniken zur Erzeugung von DNA-Vektoren (d.h. Trägern) für die Unterweise der Einschleppung fremder DNA in das Genom des Tieres. Diese Vektoren ermöglichen die Ausbreitung ausgewählter Gene und deren entsprechende Modifikation, abhängig von der Art der Forschung, die durchgeführt wird. Der Begriff "transgenes Tier" wurde von Gordon und Ruddle3eingeführt.
Die erste weithin akzeptierte Art, die in der Neurobiologie, Physiologie, Pharmakologie, Toxikologie und vielen anderen Bereichen der biologischen und medizinischen Wissenschaften verwendet wurde, war die Norwegische Ratte, Rattus norvegicus4. Aufgrund der Schwierigkeit, Rattenembryonen zu manipulieren, ist die Hausmaus Mus musculus jedoch die dominierende Tierart in der Genforschung geworden5. Ein weiterer Grund für den Vorrang der Maus in solchen Forschungen war die Verfügbarkeit embryonaler Stammzelltechnologie, um Knockout-Tiere für diese Art zu erzeugen. Die am häufigsten verwendete Technik der Transgenese (2–10% der transgenen Nachkommen im Vergleich zu allen geborenen Tieren) ist die Mikroinjektion von DNA-Fragmenten in einen Pronukle einer befruchteten Oozyte. 1990 wurde dieser Ansatz, der erstmals bei Mäusen eingeführt wurde, für Ratten6,7angepasst. Rattentransgenese durch pronukleare Injektion ist durch eine geringere Effizienz8 im Vergleich zu Mäusen gekennzeichnet, die eng mit dem Vorhandensein von elastischem Plasma und pronukleären Membranen 9 zusammenhängt.9 Obwohl das Überleben von Embryonen nach Dermanipulation um 40–50 % niedriger ist als bei Mäusen, gilt diese Technik als Standard bei der Erzeugung genetisch veränderter Ratten10. Es wurden alternative Ansätze untersucht, die eine effiziente Transgenintegration und höhere Überlebensraten von injizierten Zygoten gewährleisten können.
Der Schlüsselfaktor für eine stabile Transgenexpression und Übertragung auf Nachkommen ist seine Integration in das Wirtszellgenom. Lentiviren (LVs) haben das Unterscheidungsmerkmal, dass sie sowohl teilende als auch nicht trennende Zellen infizieren können. Ihre Verwendung als Werkzeug für die Aufnahme heterologer Gene in Embryonen erwies sich als hocheffizient11, und transgene Individuen zeichnen sich durch eine stabile Expression des eingebauten DNA-Fragments aus. Die Wirksamkeit von lentiviralen Vektoren wurde für die genetische Veränderung von Mäusen12,13, Ratten12,14und anderen Arten11bestätigt. Bei dieser Methode wird die LV-Suspension im Stadium von zwei Vorkernen unter die Zona pellucida des Embryos injiziert. Diese Technik garantiert im Wesentlichen ein 100%-Überleben der Embryonen, da das Oolemma nicht betroffen bleibt. Entscheidend ist die Herstellung hochwertiger und relativ hochkonzentrierter LV-Aufhängungen. Niedrigere Konzentrationen von LV-Suspensionen können jedoch durch wiederholte Injektionen überwunden werden11, was die Menge an Viruspartikeln an der Eioberfläche erhöht, ohne die Membranintegration zu beeinträchtigen. Embryonen, die wiederholten Injektionen in den Perivitelline-Raum ausgesetzt sind, entwickeln sich weiter, und transgene Nachkommen können das Transgen durch die Keimbahn übertragen. Die Effizienz der transgenen Rattenerzeugung durch lentivirale Transgenese kann bis zu 80%12betragen.
Hier beschreiben wir die Produktion von HIV-1-abgeleitetem rekombinantem Lentivirus, das pseudotypisiert mit vesikulärem Stomatitisvirus (VSV) G-Hüllprotein war. Der Einsatz des Verpackungssystems VSV-Pseudotyp der zweiten Generation bestimmt die große Infektiosität von Viruspartikeln und ermöglicht die Produktion hochstabiler Vektoren, die durch Ultrazentrifugation und Kryopreservede konzentriert werden können. Nach der Titer-Verifizierung sind die Vektoren bereit, als Vehikel für die Transgenabgabe in Albino Wistar Rattenzygoten verwendet zu werden. Nach einer Reihe von Injektionen können die Embryonen über Nacht kultiviert und im Zwei-Zellen-Stadium an Pflegemütter übertragen werden. An dieser Stelle kann einer von zwei alternativen Ansätzen in Betracht gezogen werden. Das Standardverfahren verwendet pseudoschwangere Weibchen als Embryo-Empfänger. Wenn die Schwangerschaftsrate nach der Paarung mit vasectomisierten Männchen niedrig ist, können die Embryonen jedoch in schwangere Wistar/Sprague-Dawley (SD) Weibchen implantiert werden, die mit fruchtbaren männlichen Ratten mit einer dunklen Fellfarbe gepaart sind (z. B. Brown Norway [BN] Ratten). Die Farbe des Fells ermöglicht die Unterscheidung von Nachkommen von natürlicher Schwangerschaft von Nachkommen, die aus den übertragenen manipulierten Embryonen stammen.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Die Herstellung und Anwendung von Virusvektoren entsprach den Richtlinien der Biosicherheitsstufe 2 und wurde vom polnischen Umweltministerium genehmigt. Alle versuchsweisen Tierverfahren, die unten beschrieben werden, wurden von der Lokalen Ethikkommission genehmigt. Die Tiere wurden in individuell belüfteten Käfigen bei stabiler Temperatur (21–23 °C) und Luftfeuchtigkeit (50–60%) untergebracht. mit ad libitum Zugang zu Wasser und Nahrung unter einem 12 h/12 h Licht/Dunkel-Zyklus.
1. Lentivirale Vektorproduktion
- Transfektion von HEK 293T-Zellen
HINWEIS: Das hier vorgestellte Protokoll ist für die Transfektion von zwanzig bis 10 cm großen Kulturgerichten konzipiert, die etwa 200 ml rohen Vektorüberstand esen lassen.- Kultur HEK 293T-Zellen in DMEM-Medium, das mit fetalem Rinderserum (10%,2 v/v) in einem befeuchteten CO2-Inkubator bei 37 °C ergänzt wird. Für die Transfektion 20 10 cm Durchmesser Platten vorbereiten, und Samen 1,5–2 x 106 HEK 293T Zellen pro Schale.
- Wenn der Zusammenfluss 70 % erreicht, transfeken Sie die Zellen mit Polyethylenimin (PEI)-Reagenz, pH 7,0, im Verhältnis von 3 g PEI pro 1 g DNA.
- Bereiten Sie die Transfektionsmischung für fünf Teller vor (bereiten Sie die Anzahl der Wiederholungen entsprechend der Gesamtzahl der Gerichte). Fügen Sie die Mischung aus drei Plasmiden auf 1 ml des Modifizierten Eagle Mediums (DMEM; ohne Serum) von Dulbecco hinzu, so dass sie eine Endmenge von 25 g VSVg-Plasmid, 50 g Delta R8.2 und 50 g kodierender Plasmid erreichen.
- Pipette nach oben und unten, und fügen Sie 125 l PEI in einer Konzentration von 3 g / l. Inkubieren bei Raumtemperatur für 15 min, invertieren sie das Rohr dreimal während der Inkubation. Fügen Sie 200 l des Transfektionsgemisches pro Platte hinzu. Als nächstes inkubieren Sie die2 Platten in einem befeuchteten CO2-Inkubator bei 37 °C.
- Konzentration von lentiviralen Vektoren
- Achtundvierzig Stunden nach der Transfektion ernten Sie das Medium, das LV-Partikel enthält. Verwenden Sie 50 ml konische Rohre.
HINWEIS: Bei Verwendung eines Plasmids mit einem fluoreszierenden Tag können Zellen an dieser Stelle visualisiert werden, um die Transfektionseffizienz zu überprüfen. Ein neuer Teil des DMEM-Mediums kann hinzugefügt werden, und Zellen können für weitere 24 h inkubiert werden. Die LV-Ausbeute ist vergleichbar, wenn sie bei den 48 und 72 h-Zeitpunkten nach der Transfektion gesammelt wird. - Zentrifugieren Sie das Medium bei 3.000 x g für 5 min und Raumtemperatur, um abgetrennte Zellen zu entfernen.
- Filtern Sie den Überstand (0,45 m) und gießen Sie ihn in neue Schläuche.
HINWEIS: Dieser Schritt kann weggelassen werden. - DNase I (RNase-frei, 1 g/ml) und MgCl2 (1 mM) hinzufügen und in einem Wasserbad bei 37 °C für 15 min inkubieren.
- Übertragen Sie die mittleren zu Einweg-Polyethylen-Rohre und Ultrazentrifuge in einem schwingenden Rotor bei 115.000 x g und 4 °C für 1,5 h.
- Nach dem Zentrieren die Wände der Rohre vorsichtig von den mediumRückständen abtropfen lassen.
- Das Pellet mit steriler Phosphat-gepufferter Saline (PBS; 70–80 l pro Tube) einweichen.
- 2 h bei 4–8 °C inkubieren.
- Setzen Sie die viralen Vektoren in PBS durch sanftepipettierende Übertragung endiel.
VORSICHT: Vermeiden Sie Schaumbildung. - Auf ein 1,5 ml Zentrifugenrohr und eine Zentrifuge bei 7.000 x g und 4 °C für 30 s übertragen. Überleitung des Überstandes auf ein neues Rohr übertragen. Wiederholen Sie diesen Schritt, bis kein zelluläres Schmutzpellet sichtbar ist.
- Aliquot und einfrieren bei -80 °C. Vermeiden Sie das erneute Einfrieren des LV aliquot.
- Achtundvierzig Stunden nach der Transfektion ernten Sie das Medium, das LV-Partikel enthält. Verwenden Sie 50 ml konische Rohre.
- Bestimmung des Virustiters mittels quantitativer Polymerase-Kettenreaktion
HINWEIS: Die Titration viraler Vektoren erfolgt mit quantitativer PCR (qPCR). Diese Methode basiert auf der Verstärkung eines doppelsträngigen 84 bp langen DNA-Fragments innerhalb des langen terminalen Repeat-Bereichs des viralen Genoms15.- Bereiten Sie die Standardkurve vor, indem Sie serielle Verdünnungen des LV-Kodierungsplasmids erstellen: 1:500, 1:1.000, 1:5.000, 1:10.000, 1:100.000 und 1:1.000.000. Bestimmen Sie die Anzahl der Kopien des Plasmids, die für die Standardkurve verwendet werden. Verwenden Sie die folgende Formel: Anzahl der Kopien/ L = (Konzentration [g/l] x 6,02 x 1023 [Zahl/mol]) / (660 [g/mol] x Plasmidgröße [bp]), wobei 6,02 x 1023 Zahl/Mol die Zahl von Avogadro und 660 g/mol das bp-Gewicht ist.
HINWEIS: Online-Kopiernummernrechner können verwendet werden. - Vorbereiten von Verdünnungen der lentiviralen Suspension: 1:100, 1:500 und 1:1.000.
- Bereiten Sie das Reaktionsgemisch (Volumen pro Bohrung): 10 l qPCR Mastermix, 1 l von 10 'M Vorwärtsprimer, 1 l von 10 'M Reverse Primer und 7 'L von H2O. Pipette das Gemisch in die Bohrungen von 96-Well-Platten.
HINWEIS: Vorwärtsgrundierung: 5'-AGCTTGCCTTGAGTGCTTCA. Reverse Primer: 5'-TGACTAAAAGGGTCTGAGGGA. - Fügen Sie 1 l jeder Standardverdünnung und lentiviralen Suspension in Dreifachersäure hinzu.
- Führen Sie die qPCR nach folgenden Parametern aus: 50 °C für 2 min, 96 °C für 5 min und 35 Zyklen von 96 °C für 20 s, 60 °C für 40 s und 70 °C für 1 min, gefolgt von Schmelzkurvenstufe: 95 °C für 1 min und 60 °C bei 30 s.
- Analysieren Sie die Ergebnisse, indem Sie die Anzahl der Moleküle, die für jede Verdünnung empfangen werden, mit der Standardkurve vergleichen. Bestimmen Sie die Konzentration von Vektormolekülen als Durchschnitt von drei Replikationen für jede Verdünnung.
HINWEIS: Die vorgestellte Quantifizierung gibt die physikalische Konzentration der virusischen Partikel an. Es sollte nicht als funktioneller Titer behandelt werden.
- Bereiten Sie die Standardkurve vor, indem Sie serielle Verdünnungen des LV-Kodierungsplasmids erstellen: 1:500, 1:1.000, 1:5.000, 1:10.000, 1:100.000 und 1:1.000.000. Bestimmen Sie die Anzahl der Kopien des Plasmids, die für die Standardkurve verwendet werden. Verwenden Sie die folgende Formel: Anzahl der Kopien/ L = (Konzentration [g/l] x 6,02 x 1023 [Zahl/mol]) / (660 [g/mol] x Plasmidgröße [bp]), wobei 6,02 x 1023 Zahl/Mol die Zahl von Avogadro und 660 g/mol das bp-Gewicht ist.
2. Erzeugung von transgenen Ratten
- Superovulation und Sammlung befruchteter Embryonen
- Gonadotropine verabreichen.
HINWEIS: Um die Anzahl der gesammelten Embryonen (ca. 30 pro Weibchen) zu erhöhen, verwenden Sie unreife 5 Wochen alte Wistar-Weibchen zur hormonellen Stimulation.- Am 1. Tag (12–13 Uhr) injizieren sie das Serum gonadotropin der trächtigen Stute (PMSG; 25 I.E. pro Weibchen). Bereiten Sie 1 ml Aliquots der Arbeitslösung in einer Konzentration von 125 I.E./ml vor, indem Sie Hormonpulver in 0,9% NaCl auflösen. Bei -20 °C bis zu 1 Monat oder -80 °C für bis zu 6 Monate lagern.
- Am 3. Tag (12.00–10.00 Uhr) wird das menschliche Choriongonadotrophin (hCG; 30 I.E. pro Weibchen) intraperitoneal injiziert. Bereiten Sie 1 ml Aliquots der Arbeitslösung (150 I.E./ml) durch Auflösen von Hormonpulver in 0,9% NaCl vor. Bei -20 °C bis zu 1 Monat oder -80 °C für bis zu 6 Monate lagern.
- Nach hCG-Verabreichung paaren Weibchen 1:1 mit sexuell fruchtbaren Männchen (3-10 Monate alt).
- Am nächsten Morgen (Tag 4 um 8–10 Uhr) überprüfen Sie die Weibchen auf das Vorhandensein eines Vaginalsteckers. Überprüfen Sie die vaginale Öffnung auf das Vorhandensein eines weißlichen Steckkerzes, der für die beste Visualisierung am frühen Morgen nach der Paarungsnacht überprüft werden sollte. Für die Embryo-Sammlung verwenden Sie nur Weibchen mit einem sichtbaren Stecker.
- Sammeln Sie Embryonen um 10 Uhr. Opfern Sie die Tiere, um die Eileiter zu verbrauchen, und sammeln Sie die Eileiter in einer Schüssel mit vorgewärmten M2 Medium.
- Übertragen Sie die Eileiter auf eine 35-mm-Schale, die vorgewärmtes M2-Medium mit Hyaluronidase aus Rinderhoden in einer Konzentration von 0,5 mg/ml enthält.
- Öffnen Sie die Wände des Eileiters mit feiner Zange unter einem Stereomikroskop und drücken Sie die Ampulla (d.h. den geschwollenen Teil des Eileiters, der befruchtete Embryonen enthält, die von Cumuluszellen umgeben sind), bis die Embryonen befreit sind.
HINWEIS: Hyaluronidase verdaut Zellvon- und Zellerzellen enzymatisch und setzt Embryonen frei.
VORSICHT: Eine längere Exposition gegenüber Hyaluronidase ist schädlich für Embryonen; Daher sollte dieser Schritt nicht länger als 5 min dauern. - Um die Freisetzung von Embryonen aus Cumuluszellen zu erleichtern, pipetten Sie sie vorsichtig mit einer Glastransferpipette, die mit einem mundbetätigten Saugrohr verbunden ist.
- Um die Transferpipette zu produzieren, ziehen Sie eine glasige Pasteurpipette über eine Flamme, um eine gerade Spitze von 5-10 cm zu erzeugen. Brechen Sie die Pipette und hinterlassen Sie eine Spitze von 4 cm.
- Waschen Sie die Embryonen ein paar Mal in M2 Medium, um Hyaluronidase und zelluläre Ablagerungen zu entfernen. Übertragen Sie die Embryonen in eine 60-mm-Schale, die Tropfen vorädglichem M16-Medium, bedeckt mit flüssigem Paraffin oder Mineralöl, in einem befeuchteten 37 °C-Inkubator mit einerCO2-Atmosphäre von 5 % enthält.
- Gonadotropine verabreichen.
- Mikroinjektion von lentiviralen Vektoren in einzelligen Embryo unter der Zona pellucida
HINWEIS: Verwenden Sie einzellige Embryonen mit zwei sichtbaren Vorkernen für die Mikroinjektion (Abbildung 1).- Das LV aliquot bei Raumtemperatur und Zentrifuge bei 10.000 x g und RT für 2 min auftauen, um restliche Zellreste zu pellet.
- Microinjection-Setup
- Bereiten Sie Glashaltepipetten (Borosilikatglaskapillare) mit einer Mikroschmiede vor. Ziehen Sie die Glaskapillare über eine Flamme, um eine 5–10 cm Spitze zu erzeugen. Brechen Sie die Pipette und hinterlassen Sie eine Spitze von 4 cm. Der Außendurchmesser sollte 80–120 m betragen.
HINWEIS: Stellen Sie sicher, dass die Pipettenspitze perfekt gerade und glatt ist. - Montieren Sie die gezogene Pipette in einer Mikroschmiede mit der Spitze vor dem Heizfilament. Erhitzen Sie das Filament sehr nahe an der Pipettenspitze und lassen Sie es auf einen Durchmesser von 15 m schrumpfen (ca. 20% der Embryogröße). Positionieren Sie die Pipette senkrecht zum Heizfilament, 2–3 mm von der Pipettenspitze entfernt, und beginnen Sie zu erwärmen. Das Glas wird weicher. Erhitzen, bis es einen 15°-Winkel erreicht.
- Bereiten Sie Mikroinjektion Borosilikat Glas kapillaren mit einem Filament mit einem Pipettenzieher. Setzen Sie die Kapillare in die Zugkammer ein. Führen Sie einen Rampentest durch (erstmals für neues Glas und jedes Mal nach dem Wechseln des Filaments). Stellen Sie die Hitze auf den Rampenwert -10, Ziehen sie auf 100, die Geschwindigkeit auf 150 und die Zeit auf 100.
HINWEIS: Ändern Sie die Parameter, um eine optimale Injektionskapillare zu erhalten. - Unter einer biosafety laminaren Durchflusshaube laden Sie ca. 2 l der virusfreien Lösung mit einer Mikroladerspitze in die Mikroinjektionspipette.
- Bereiten Sie eine Mikroinjektionsschale (Deckel von 60 mm Petrischale) mit einem 100 l Tropfen M2 Medium (in der Mitte), bedeckt mit flüssigem Paraffin oder Mineralöl.
- Montieren Sie die Haltepipette und Die Mikroinjektionskapillare, die mit viraler Lösung beladen ist, unter einem invertierten Mikroskop mit einer Mikromanipulator- und Mikroinjektionsschale.
- Bereiten Sie Glashaltepipetten (Borosilikatglaskapillare) mit einer Mikroschmiede vor. Ziehen Sie die Glaskapillare über eine Flamme, um eine 5–10 cm Spitze zu erzeugen. Brechen Sie die Pipette und hinterlassen Sie eine Spitze von 4 cm. Der Außendurchmesser sollte 80–120 m betragen.
- Führen Sie die Mikroinjektion durch.
- Übertragen Sie 15–20 einzellige Embryonen auf den M2-Tropfen auf der Mikroinjektionsschale. Halten Sie den Embryo mit einer Haltepipette.
- Mit 400-facher Vergrößerung injizieren Sie die LV-Lösung unter der Zona pellucida mit der Glaskapillare, die mit einem automatischen Injektor verbunden ist, in den Perivitelline-Raum. Halten Sie die Kapillare unter der Zona pellucida für einen Moment.
HINWEIS: Mit sanftem Gegendruck fließt die virusvirale Lösung kontinuierlich aus der Injektionskapillare, aber das Volumen der suspension, die geliefert wird, kann nicht kontrolliert werden. - Mit einer feinen Pipette die Embryonen bei 37 °C in einer 5%-CO2-Atmosphäre in die Kulturschale im Brutkasten zurückbringen.2 Die Anzahl der Injektionen einer Zygote kann variieren und kann basierend auf der viralen Vektorkonzentration angepasst werden.
HINWEIS: Die injizierten Embryonen können im Einzellstadium oder inkubiertem O/N in M16-Medium an Pflegemütter übertragen werden, bevor sie im Zweizellstadium übertragen werden. Längere In-vitro-Kultur von Rattenembryonen sollte vermieden werden.
- Transfer von injizierten Embryonen an Pflegemütter
- Bereiten Sie Pflegemütter vor, indem Sie geschlechtsreife SD-Weibchen mit fruchtbaren BN-Männchen oder mit vasectomisierten SD-Männchen (das Vasektomie-Verfahren wird in Abschnitt 3 unten beschrieben) am Tag 3 (für die Übertragung von Embryonen im Einzellstadium) oder Tag 4 (für die Übertragung von Embryonen im Zwei-Zell-Stadium) paaren.
HINWEIS: Für die Oviduct-Übertragung verwenden Sie 0,5 Tage nach coitum (dpc) Weibchen. - Überprüfen Sie am nächsten Morgen SD-Weibchen auf einen Vaginalstecker, und verwenden Sie nur solche mit einem sichtbaren Stecker.
- Embryotransfer durchführen.
HINWEIS: Führen Sie den chirurgischen Eingriff mit sterilen Instrumenten unter einem Stereomikroskop durch. Vor dem Tag der Operation, AutoklavSchere, feine Zange, Nadelhalter und Skalpellhalter.- Anästhetisieren Sie ein Weibchen mit i.p. Verabreichung von Ketamin (50 mg/kg) und Medetomidin (0,5 mg/kg) Lösung. Testen Sie auf Reflexe, um die Anästhesie zu bestätigen, bevor Sie mit dem chirurgischen Eingriff beginnen.
- Injizieren Sie das Tier subkutan mit Tolefenaminsäure (2 mg/kg), Butorphanoltartrat (1 mg/kg) und Enrofloksacin (5–10 mg/kg), um Entzündungen, Schmerzen und Infektionen zu verhindern.
- Tragen Sie die ophthalmologische Salbeschmierung auf beide Augen auf, um eine Hornhauttrocknung zu verhindern. Rasieren Sie das Fell von der Rückseite, und sterilisieren Sie die Haut mit chirurgischem Peeling gefolgt von 70% Alkohol mit sterilen nicht haftenden Pads. Lassen Sie die Haut trocknen.
- Injizieren Sie das Tier subkutan mit 100 l 0,25% Bupivacain (Lokalanästhetikum) an der Einschnittstelle. Übertragen Sie das Tier in einer anfälligen Position auf eine saubere Oberfläche auf einem Heizkissen unter dem Ziel eines chirurgischen Mikroskops. Bedecken Sie die Ratte mit einem sterilen Vorhang mit einem kleinen Loch, das über den unteren Rücken geschnitten wird.
- Führen Sie einen ca. 2 cm langen Hautschnitt parallel zur Lendenwirbelsäule durch.
- Mit einer scharfen Schere, machen Sie einen Schnitt in der Bauchwand. Schnappen Sie sich ein Eierstockfettpad mit Zangen, ziehen Sie den Eierstock und das Eileiter heraus und legen Sie sie auf Gaze, die mit 0,9% NaCl benetzt wird.
- Aspirieren Sie M2 medium, drei Luftblasen und die Embryonen in die Transferkapillare. Empfohlene Gesamtzahl der zu übertragenden Embryonen (einseitig oder bilateral): schwangere Weibchen (ca. 15–16 Embryonen), pseudoschwangere Weibchen (ca. 30 Embryonen).
- Machen Sie einen kleinen Schnitt im Eileiter (zwischen infundibulum und ampulla) mit Mikroschere, und legen Sie die Transferpipette in das Eileiter.
- Tragen Sie Embryonen und Luftblasen vorsichtig von der Pipette zum Eileiter. Mit stumpfer Zange, legen Sie den Fortpflanzungstrakt wieder in die Bauchhöhle.
- Die Bauchwand mit polyglykolsäureabsorbierbaren Nähten bedecken und den Hautschnitt mit Wundclips schließen. Abhängig von der Anzahl der verfügbaren Embryonen, wiederholen Sie dieses Verfahren für das andere Eileiter.
- Injizieren Sie das Tier intraperitoneal mit Atipamezol (0,5 mg/kg), um die Wirkung der Anästhesie umzukehren.
- Übertragen Sie das Tier in einen sauberen Käfig und halten Sie es auf einer wärmenden Platte, um sich vollständig von der Anästhesie zu erholen. Die Lieferung bei Ratten erfolgt nach 21 Tagen.
HINWEIS: Wenn männliche BN-Ratten zur Paarung verwendet werden, sind nur weiße Welpen potenziell transgen; braune Welpen stammen aus natürlicher Schwangerschaft. - Sammeln Sie Gewebefragmente (vorzugsweise vom Ohr) zu Genotyp 3 Wochen alten Welpen.
- Bereiten Sie Pflegemütter vor, indem Sie geschlechtsreife SD-Weibchen mit fruchtbaren BN-Männchen oder mit vasectomisierten SD-Männchen (das Vasektomie-Verfahren wird in Abschnitt 3 unten beschrieben) am Tag 3 (für die Übertragung von Embryonen im Einzellstadium) oder Tag 4 (für die Übertragung von Embryonen im Zwei-Zell-Stadium) paaren.
3. Vasektomie
HINWEIS: Vor dem Tag der Operation, AutoklavSchere, feine Zange und Nadelhalter.
- Anästhetisieren Sie eine 5 Wochen alte männliche SD-Ratte mit i.p. Verabreichung von Ketamin (50 mg/kg) und Medetomidin (0,5 mg/kg) Lösung. Testen Sie auf Reflexe, um die Anästhesie zu bestätigen, bevor Sie mit dem chirurgischen Eingriff beginnen.
- Tolfenamsäure (2 mg/kg), Butorphanoltartrat (1 mg/kg) und Enrofloksacin (5–10 mg/kg) subkutan verabreichen, um Entzündungen, Schmerzen bzw. Infektionen zu verhindern.
- Tragen Sie die ophthalmologische Salbeschmierung auf beide Augen auf, um eine Hornhauttrocknung zu verhindern. Legen Sie die Ratte auf eine saubere Oberfläche auf einem Heizpad, und sterilisieren Sie die Haut auf den Hoden mit chirurgischem Peeling gefolgt von 70% Alkohol mit sterilen nicht haftenden Pads. Lassen Sie die Haut trocknen. Bedecken Sie die Ratte mit einem sterilen Vorhang mit einem kleinen Loch, das über die Hoden geschnitten wird. Drücken Sie vorsichtig den Bauch, um die Hoden im Skrotalsack freizulegen.
- Mit einer chirurgischen Schere, machen Sie einen Schnitt von 0,5 cm in der Mitte des Skrotalsacks. Suchen Sie die Mittellinie (weißliche Linie) zwischen den Hoden.
- Machen Sie einen 5 mm Schnitt in der Hodenmembran in der Nähe der linken Seite der Mittellinie Wand.
- Schieben Sie den Hoden vorsichtig nach links und lokalisieren Sie Vas deferens (zwischen Hoden und Mittellinie) als weißen Kanal mit einem einzigen Blutgefäß.
- Ziehen Sie die Vasen vorsichtig mit der Zange eines Uhrmachers aus dem Skrotalsack. Halten Sie die Vas deferens mit einem Paar Zangen, und schneiden Sie es mit feiner Schere (oder kauterisieren mit rot-heißen Spitzen eines zweiten Paares von Zangen). Entfernen Sie ein 1 cm großes Fragment des Kanals.
HINWEIS: Wenn eine Kauterisierung durchgeführt wird, halten Sie die Spitze des zweiten Zangenpaares in der Flamme. - Wiederholen Sie das obige Verfahren für die anderen Hoden. Die Haut mit polyglykolsäureregetisierbaren Nähten bedecken und das Tier intraperitoneal mit Atipamezol (0,5 mg/kg) injizieren.
- Legen Sie die Ratte in einen sauberen Käfig auf eine wärmende Platte, bis sich das Tier von der Anästhesie erholt.
HINWEIS: Männchen können in den Testpaarungen nach einer 2-wöchigen Erholungsphase verwendet werden. Nachdem die Sterilität bestätigt wurde, können sie für die Pseudoschwangerschaftsinduktion verwendet werden.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Unter Verwendung des hier beschriebenen Protokolls wurden lentivirale Vektoren hergestellt, die das Syn-TDP-43-eGFP-Konstrukt trugen (physikalischer LV-Titer = 3,4 x 108/L) und konnten dann für subzonale Injektionen im Einzellstadium des Embryos verwendet werden. Nur Embryonen mit zwei sichtbaren Vorkernen wurden dem Verfahren unterzogen. Die Anzahl der Injektionen von viralen Suspensionen wurde experimentell bestimmt. Eine hohe Implantationseffizienz und ein gleichzeitiger Mangel an transgenen Nachkommen wurden als Indikatoren für eine unzureichende Anzahl von Viruspartikeln für eine erfolgreiche Transduktion betrachtet. In diesem Fall wurde die Anzahl der Injektionen erhöht. Die einzige Verabreichung von LV führte zur Geburt von 20 Ratten der F0-Generation, von denen keine transgene war. Eine Erhöhung der Anzahl der Injektionen um eine Größenordnung resultiert nicht in der Geburt von Ratten, sondern 100% der Embryonen entwickelten sich bis in das Zwei-Zell-Stadium. In den folgenden Experimenten wurde die Anzahl der Injektionen um eins im Vergleich zu dem Wert erhöht, für den die Nachkommen erhalten wurden. Für die Variante von zwei Injektionen wurden acht Ratten geboren, von denen drei bestätigt wurden, um das Transgen zu tragen (in Tabelle 1zusammengefasst). Einer der Gründer übertrug das Transgen nicht auf den Nachwuchs. Die Anzahl der Embryonen, die in jeder experimentellen Variante injiziert und übertragen wurden, betrug 48 in den Varianten LV x1 und LV x2 und 45 in LV x10. Für jedes Versuchsaufbau wurden drei Pflegeweibchen eingesetzt. Der gewählte Ansatz ermöglichte die Erzeugung stabiler transgener Rattenlinien, die das TDP-43-eGFP-Fusionsprotein unter Kontrolle des neuronalen Synapsin-1-Promotors im gesamten zentralen Nervensystem ausdrückten (Abbildung 2A,B)14. Die Lentivirus-basierte Transgenese führte zu einer einzigen Kopie des Transgens, wie qPCR (Abbildung 2C) zeigt.
In dem oben beschriebenen Versuchsaufbau betrug die Überlebensrate der injizierten Embryonen 95 %. Ähnliche Ergebnisse wurden erzielt, wenn die gleiche Methode für andere lentivirale Vektoren verwendet wurde, wie in Tabelle 2zusammengefasst. Der Anteil der Embryonen, die die pronukleären Injektionen überlebten, war deutlich geringer (29–45%). Tabelle 2 fasst die repräsentativen Ergebnisse der Implantationseffizienz manipulierter Zygoten unter Berücksichtigung der Übertragung von pseudoschwangeren vs. schwangeren Frauen zusammen. Die Verwendung von nicht manipulierten Embryonen zusammen mit injizierten Embryonen wurde zuvor berichtet16. Unsere Gesamtergebnisse deuten darauf hin, dass schwangere weibliche Ratten als Pflegemütter mit vergleichbarer Effizienz verwendet werden können. Wir erhielten einen ähnlichen Prozentsatz der Implantation von ausländischen Embryonen bei schwangeren und pseudoschwangeren Ratten (Gesamtdurchschnitt für mehrere Versuchseinrichtungen: 15% vs. 16%). Die Implantationsrate war jedoch höher, wenn die Embryonen einer subtileren Manipulation unterzogen wurden, was eine subzonale Injektion bedeutet (10% vs. 21%). Insbesondere zeigten die numerischen Daten, die für einzelne Mikroinjektionsrunden analysiert wurden, dass die Wirksamkeit der Implantation von der Anzahl der Injektionen eines Embryos abhing(Tabelle 1,letzte Spalte) und indirekt von der Viruslast abhing.
| Vektor | Anzahl der Injektionen/Embryo | Anzahl der injizierten Embryonen | Anzahl der Welpen | Anzahl pflegemütter | Anzahl der transgenen Gründer | Implantationseffizienz für jede Variante |
| Syn-TDP-43WTLV | 1 | 48 | 20 | 3 | 0 | 42% |
| 10 | 45 | 0 | 3 | 0 | 0% | |
| 2 | 48 | 8 | 3 | 3 | 17% |
Tabelle 1: Zusammenfassung der Anzahl der subzonalen Injektionen von Zygoten mit Syn-TDP-43WTlentiviralen Vektoren.
| Methode | Vektor | Titer/Konzentration | Anzahl der injizierten Embryonen | Überlebte Embryonen | Überlebensrate | Zahl der Pflegemütter | Anzahl der Welpen | Implantationseffizienz | Schwangerschaft (P) /Pseudoschwangerschaft (PP) |
| Pni | TTYH1-Thy1-EGFP | 1 ng/l | 1083 | 424 | 39% | 16 | 54 | 13% | Pp |
| Pni | H3mCherry | 0,5-2 ng/l | 2229 | 647 | 29% | 29 | 67 | 10% | Pp |
| Pni | Syn-TDP-43-A315T | 2 ng/l | 1256 | 562 | 45% | 31 | 42 | 7% | Pp |
| Lv | Syn-TDP-43-A315T | 8,7 x 108 | 115 | 106 | 92% | 7 | 18 | 17% | P |
| Lv | Syn-TDP-43 WT | 3,4 x 108 | 152 | 141 | 93% | 9 | 28 | 20% | P |
| Lv | LVH3mcherry | 1,3 x 107 | 504 | 450 | 89% | 13 | 115 | 26% | Pp |
Tabelle 2: Überlebensrate des Embryos und Implantationseffizienz, abhängig von der verwendeten Injektionsmethode und Schwangerschaft versus Pseudoschwangerschaftsinduktion. PNI, pronukleare Injektion; LV, lentivirale Vektor subzonale Injektion.

Abbildung 1: Mikroskopische Aufnahme eines einzelligen Rattenembryons, das für die subzonale lentivirale Vektorinjektion vorbereitet wurde. Der Embryo wurde mit einer Haltepipette immobilisiert. Zwei Vorkerne, die mütterliches und väterliches genetisches Material und den polaren Körper enthielten, sind sichtbar. Skalenbalken = 20 m. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
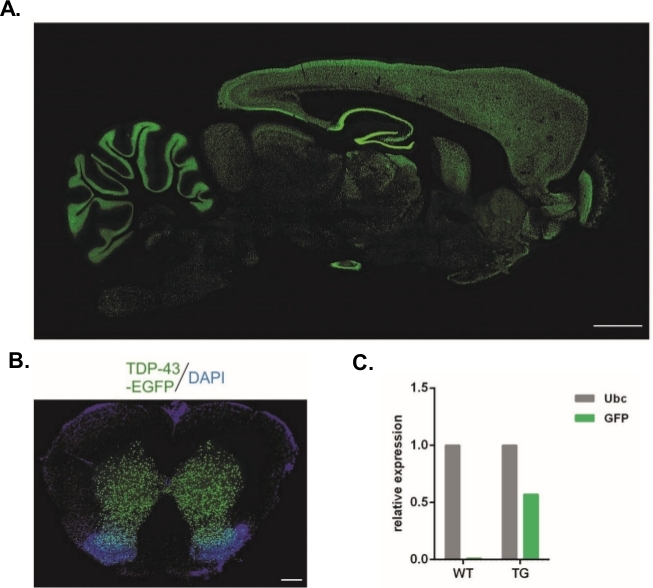
Abbildung 2: Erzeugung stabiler transgener Rattenlinien, die das TDP-43-eGFP-Fusionsprotein unter Kontrolle des neuronalen Synapsin-1-Promotors im gesamten zentralnervösen System ausdrückten. (A) Synapsin-1 (Syn)-gesteuertes hTDP-43-eGFP-Expressionsmuster in einem sagittalen Abschnitt des transgenen Rattenhirns. Skala bar = 3 mm. (B) Koronaler Abschnitt des Rückenmarks einer transgenen Ratte, wo eGFP Fluoreszenz, mit DAPI kontragefärbt, auf graue Substanz des Rückenmarks beschränkt war. Skalenbalken = 250 m. (C) Relative Expression des GFP-Transgentranskripts im Vergleich zum Ubiquitin-C-Referenztranskript. n = 2 Wildtyp. n = 2 transgene. Die Zahl wurde von14geändert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Fortschritte in den transgenen Technologien haben Nagetiermodelle zu einem unschätzbaren Werkzeug in der biomedizinischen Forschung gemacht. Sie bieten die Möglichkeit, Genotyp-Phänotyp-Beziehungen in vivo zu untersuchen. Hier stellen wir eine weit verbreitete Alternative zur konventionellen Transgenese durch pronukleäre Injektionen vor. Die Verwendung von lentiviraler Gentransduktion umgeht die Notwendigkeit anspruchsvoller Mikroinjektionen, da virale Vektoren unter der Zona pellucida injiziert werden können. Dieser Ansatz hat keinen Einfluss auf die Integrität des Embryos, was im Wesentlichen eine Überlebensrate von 100 % für injizierte Zygoten garantiert. Das Transgen, das mittels lentiviraler Vektoren eingearbeitet wird, ist stabil in das Wirtsgenom integriert, was eine langfristige Expression und Keimbahnübertragung ermöglicht. Darüber hinaus stellen wir Pflegemüttern zwei alternative Techniken für den veränderten Embryotransfer vor. Eine Technik nutzt embryonale Übertragung auf pseudoschwangere Weibchen, die zuvor durch Paarung mit vasectomisierten unfruchtbaren Männchen hergestellt wurden. Die andere Technik basiert auf der Verwendung von natürlich schwangeren Weibchen, die mit fruchtbaren Männchen, aber mit einer anderen Fellfarbe (d.h. BN-Ratten) gepaart sind. Dieser physiologischere Verlauf der Schwangerschaft ermöglicht die richtige Entwicklung von Embryonen, die herausfordernde genetische Veränderungen durchlaufen16.
Die ersten erfolgreichen Versuche, transgene Ratten zu erzeugen, wurden 1990 gemeldet7. Aufgrund von Schwierigkeiten bei der Rattentransgenese17ist jedoch in den letzten Jahrzehnten eine relativ geringe Anzahl transgener Rattenlinien entstanden9. Mehrere Hauptunterschiede zwischen Maus- und Rattentransgenese werden mit Mikroinjektionen beobachtet. Bei Ratten werden hauptsächlich ausgezüchtete Linien (z.B. Wistar und SD) für die Transgenese verwendet. Bei Mäusen verwenden die Forscher hauptsächlich die F1-Kreuzung von inzuchtierten Stämmen wegen ihrer höheren Fruchtbarkeit, einer besseren Reaktion auf die hormonelle Superovulation und der relativ einfachen Entwicklung von Embryonen in vitro vom Einzellstadium bis hin zu Blastozysten18. Die Induktion der Superovulation bei Ratten ist viel weniger effizient als bei Mäusen mit Standard-PMSG/hCG-Hormonstimulation. Aus diesem Grund wurden Versuche unternommen, alternative Protokolle zur Verabreichung dieser Hormone bei Ratten zu entwickeln, die eine kontinuierliche FSH-Infusion anstelle einer einzigen PMSG-Administration verwenden19. Es hat sich jedoch gezeigt, dass die durch PMSG/hCG oder FSH/hCG verursachte Superovulation eine vergleichbare Effizienz20hat. Unserer Meinung nach ist der kritischste Faktor, der die Wirksamkeit der Superovulation beeinflusst, das Alter ausgewählter Frauen. Dennoch sollten die genauen Parameter für jede Rattensorte, jedes Labor usw. getestet werden.
Das Verfahren zur Injektion von DNA-Lösung in den Vorkern eines einzelligen Embryos ist für beide Nagetierarten ähnlich. Die Vorkerne von Rattenzygoten haben jedoch keine so regelmäßigen Formen wie bei Mäusen und sind im Zytoplasma der Zelle tendenziell schwieriger zu definieren. Darüber hinaus sind die Rattenzygotzellmembran und die pronukleare Membran elastischer und zähflüssiger, was das Einführen einer Mitglas-Mikropipette erschwert, die mit DNA-Lösung belastet ist. Diese Faktoren führen zu niedrigeren Überlebensraten von Ratteneiern nach der Mikroinjektion (31–65% vs. 80% bei Mäusen) und erklären die geringere Transgenese-Effizienz bei Ratten9. Darüber hinaus kann eine intensive, mechanische Manipulation des Embryos auch die Implantationseffizienz beeinträchtigen, die in vielen Laboratorien, einschließlich unserer, maximal 10 % erreicht. Diese relativ geringe Ausbeute wird auch nach der Implantation einer entsprechenden Anzahl von Embryonenbeobachtet 21.
Eine Methode, die die oben genannten Schwierigkeiten überwindet, ist die Infektion von einzelligen Embryonen mit Retroviren. Retroviren enthalten genetisches Material in Form von RNA, das beim Eintritt in die infizierte Zelle durch Reverse-Transkriptase des Virus in die DNA transkribiert wird. Die DNA wird dann durch die Kernporen zum Zellkern transportiert, wo sie sich in Form eines Provirus in das Genom der Zelle integriert. Lentivirale Vektoren wurden verwendet, um transgene Mäuse und Ratten zu erzeugen12,14,22. Einzellige Embryonen ohne Zona pellucida können in einer Lösung mit einem lentiviralen Vektor inkubiert werden, oder der Vektor kann unter der Zona pellucida in den Perivitelline-Raum injiziert werden. Der Hauptvorteil dieser Methode ist ihre extrem hohe Effizienz, die mehr als 80% der transgenen Nachkommen erreicht. Nach einer Infektion mit dem lentiviralen Vektor können sich viele Kopien an verschiedenen Stellen in das Zygotengenom integrieren, im Gegensatz zur Transgenesemethode durch pronukleare Mikroinjektion, bei der in der Regel eine Integrationsstelle beobachtet wird12. Bei Nachkommen des transgenen Gründers, der mit lentiviralen Vektoren hergestellt wird, werden einzelne Kopien des Transgens getrennt, die sich durch unterschiedliche Expressionsprofile des Transgens in jeder Nachkommenschaft manifestieren können. Dies kann jedoch die Wahrscheinlichkeit erhöhen, ein Subjekt mit dem gewünschten Ausdrucksprofil zu erhalten, das vom Transgen abgeleitet wird. Die Beschränkungen gelten hauptsächlich für die Größe des Transgens, die auf ca. 8 kb23begrenzt ist.
Eine weitere Schwierigkeit bei der Rattentransgenese ist die Generation von Frauen, die als Leihmütter für genetisch veränderte Embryonen dienen. Im Standardverfahren werden Weibchen mit sterilen vasectomisierten Männchen gekreuzt, um Pseudoschwangerschaft zu induzieren. Bei Ratten ist die Pseudoschwangerschaftsbewertungstechnik viel schwieriger als bei Mäusen, so dass die Stimulation mit dem Gonadotropin, der Hormonagonisten freisetzt, manchmal ein paar Tage vor der Paarung mit Männern verwendet wird. Aus diesen Gründen bieten wir im beschriebenen Protokoll zwei alternative Ansätze zur Gewinnung von Pflegemüttern an. Die Gesamtimplantationseffizienz von manipulierten Zygoten bei schwangeren oder pseudoschwangeren Weibchen ist ähnlich. Das Vorhandensein natürlicher, nicht manipulierter Embryonen zusammen mit manipulierten embryonen kann jedoch die Schwangerschaftsrate verbessern16. Obwohl der Hauptunterschied bei der Implantationsrate die Manipulationstechnik ist (d. h. PNI vs. LV, 10% vs. 20%; siehe Tabelle 2), kann die Verwendung von pseudoschwangeren Frauen als Pflegemütter für einige Experimente von Vorteil sein.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Der Autor (W.K.) hat Rechte an dem Patent "Methode zur Herstellung eines transgenen Tieres" des Patentamts der Republik Polen (Nr. P 355353; 21.03.2008).
Acknowledgments
Diese Studie wurde durch das ANIMOD-Projekt im Rahmen des Team Tech Core Facility Plus-Programms der Stiftung für polnische Wissenschaft unterstützt, das von der Europäischen Union aus dem Europäischen Fonds für regionale Entwicklung an die WK kofinanziert wurde.
Materials
| Name | Company | Catalog Number | Comments |
| 7500 Real Time PCR System | Applied Biosystems | ||
| Aerrane (isoflurane) | Baxter | FDG9623 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177-5EA | |
| Atipam 5 mg/ml | Eurovet Animal Health BV | N/A | 0.5 mg/kg |
| Baytril 25 mg/ml (enrofloksacin) | Bayer | N/A | 5-10 mg/kg |
| Borosilicate glass capillaries with filament GC100TF-15 | Harvard Apparatus Limited | 30-0039 | injection capillary |
| Bupivacaine 25 mg/ml | Advanz Pharma | N/A | 0.25% in 0.9% NaCl |
| Butomidor 10 mg/ml (butorphanol tartrate) | Orion Pharma | N/A | 1 mg/kg |
| CELLSTAR Tissue Cell Culture Dish 35-mm | Greiner Bio-One | 627160 | |
| CELLSTAR Tissue Cell Culture Dish 60-mm | Greiner Bio-One | 628160 | |
| CellTram Oil | Eppendorf | 5176 000.025 | |
| Cepetor (Medetomidine) 1 mg/ml | cp-pharma | N/A | 0.5 mg/kg |
| Chorulon, Human Chorionic Gonadotrophin | Intervet | N/A | 150 IU/ ml ml 0.9% NaCl |
| DMEM low glucose | Sigma Aldrich | D6048 | |
| DNase, RNase-free | A&A Biotechnology | 1009-100 | |
| EmbryoMax Filtered Light Mineral Oil | Sigma | ES-005-C | |
| Envelope protein coding plasmid for lentiviral vectors (VSVg plasmid) | ADDGENE | 14888 | |
| FemtoJet | Eppendorf | 4i /5252 000.013 | |
| Fetal Bovine Serum | Sigma Aldrich | F9665-500ML | |
| Folligon, Pregnant Mare’s Serum Gonadotropin | Intervet | N/A | 125 IU/ml in .9% NaCl |
| HEK 293T cells | ATCC | ATCC CRL-3216 | |
| Hyaluronidase from Bovine Testis | Sigma | H4272-30MG | 0.5 mg/ml in M2 medium |
| Inverted Microscope | Zeiss | Axiovert 200 | |
| Ketamine 100mg/ml | Biowet Pulawy | N/A | 50 mg/kg |
| Liquid Paraffin | Merck Millipore | 8042-47-5 | |
| M16 medium EmbryoMax | Sigma | MR-016-D | |
| M2 medium | Sigma | M7167 | |
| Magnesium Chloride 1M | Sigma Aldrich | 63069-100ML | |
| Microforge | Narishige | MF-900 | |
| Mineral Oil | Sigma | M8410-500ML | |
| NaCl 0.9% | POLPHARMA OTC | N/A | sterile, 5ml ampules |
| Operation microscope | Inami Ophthalmic Instruments | Deca-21 | |
| Packaging system coding plasmid for lentiviral vectors (delta R8.2 plasmid) | ADDGENE | 12263 | |
| PEI reagent (Polyethylenimine, Mw ~ 25,000,), | Polysciences, Inc | 23966-1 | |
| Penicilin-streptomycin | Sigma Aldrich | P0781-100ML | |
| Phosphate Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma Aldrich | 806552-500ML | |
| Puller | Sutter Instrument Co. | P-97 | |
| Reflex Clip Applier/Reflex Clips | World Precision Instruments | 500345/500346 | |
| Safil, polyglycolic acid, braided, coated, absorbable threads | B.Braun Surgical | 1048029 | |
| Stereomicroscope | Olympus | SZX16 | |
| Surgical Sewing Thread | B.Braun | C1048040 | |
| SYBR Green PCR Master Mix | Applied Biosystem | 4334973 | |
| Tolfedine 4% (tolfenamic acid) | Vetoquinol | N/A | 2 mg/kg |
| TransferMan NK2 | Eppendorf | N/A | |
| Trypsin EDTA solution | Sigma Aldrich | T3924-500ML | |
| Ultracentrifuge | Beckman Coulter | Optima L-100 XP | |
| VacuTip | Eppendorf | 5175108.000 | holders capillary |
| Vita-POS | Ursapharm | N/A | eye ointment |
| Warming Plate | Semic | N/A | |
| Watchmaker Forceps | VWR | 470018-868 |
References
- Lazar, J., Moreno, C., Jacob, H. J., Kwitek, A. E. Impact of genomics on research in the rat. Genome Research. 15 (12), 1717-1728 (2005).
- Tarkowski, A. K. Studies on mouse chimeras developed from eggs fused in vitro. National Cancer Institute Monographs. 11, 51-71 (1963).
- Gordon, J. W., Ruddle, F. H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 214 (4526), 1244-1246 (1981).
- Gill, T. J., Smith, G. J., Wissler, R. W., Kunz, H. W. The Rat as an Experimental Animal. Science. 245 (4915), 269-276 (1989).
- Aitman, T. J., et al. Progress and prospects in rat genetics: a community view. Nature Genetics. 40 (5), 516-522 (2008).
- Hammer, R. E., Maika, S. D., Richardson, J. A., Tang, J. P., Taurog, J. D. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 63 (5), 1099-1112 (1990).
- Mullins, J. J., Peters, J., Ganten, D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 344 (6266), 541-544 (1990).
- Menoret, S., Remy, S., Usal, C., Tesson, L., Anegon, I. Generation of Transgenic Rats by Microinjection of Short DNA Fragments. Rat Genomics: Methods and Protocols. 597, 81-92 (2010).
- Tesson, L., et al.
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: Technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Ritchie, W. A., Neil, C., King, T., Whitelaw, C. B. Transgenic embryos and mice produced from low titre lentiviral vectors. Transgenic Research. 16 (5), 661-664 (2007).
- Lois, C., Hong, E. J., Pease, S., Brown, E. J., Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295 (5556), 868-872 (2002).
- Pfeifer, A., Ikawa, M., Dayn, Y., Verma, I. M. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proceedings of the National Academy of Sciences of the United States of America. 99 (4), 2140-2145 (2002).
- Koza, P., et al. Neuronal TDP-43 depletion affects activity-dependent plasticity. Neurobiology of Disease. 130, 104499 (2019).
- Scherr, M., Battmer, K., Blomer, U., Ganser, A., Grez, M. Quantitative determination of lentiviral vector particle numbers by real-time PCR. Biotechniques. 31 (3), 520 (2001).
- Canseco, R. S., et al. Gene transfer efficiency during gestation and the influence of co-transfer of non-manipulated embryos on production of transgenic mice. Transgenic Research. 3 (1), 20-25 (1994).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Yagle, M. K., Palmiter, R. D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proceedings of the National Academy of Sciences of the United States of America. 82 (13), 4438-4442 (1985).
- Armstrong, D. T., Opavsky, M. A. Superovulation of immature rats by continuous infusion of follicle-stimulating hormone. Biology of Reproduction. 39 (3), 511-518 (1988).
- Popova, E., Krivokharchenko, A., Ganten, D., Bader, M. Comparison between PMSG- and FSH-induced superovulation for the generation of transgenic rats. Molecular Reproduction and Development. 63 (2), 177-182 (2002).
- Johnson, L. W., Moffatt, R. J., Bartol, F. F., Pinkert, C. A. Optimization of embryo transfer protocols for mice. Theriogenology. 46 (7), 1267-1276 (1996).
- van den Brandt, J., Wang, D., Kwon, S. H., Heinkelein, M., Reichardt, H. M. Lentivirally generated eGFP-transgenic rats allow efficient cell tracking in vivo. Genesis. 39 (2), 94-99 (2004).
- Remy, S., et al. The Use of Lentiviral Vectors to Obtain Transgenic Rats. Rat Genomics: Methods and Protocols. 597, 109-125 (2010).
